

EGFR mediates hyperlipidemia-induced renal injury via regulating inflammation and oxidative stress: the detrimental role and mechanism of EGFR activation
- PMID: 27014908
- PMCID: PMC5029707
- DOI: 10.18632/oncotarget.8222
EGFR mediates hyperlipidemia-induced renal injury via regulating inflammation and oxidative stress: the detrimental role and mechanism of EGFR activation
Abstract
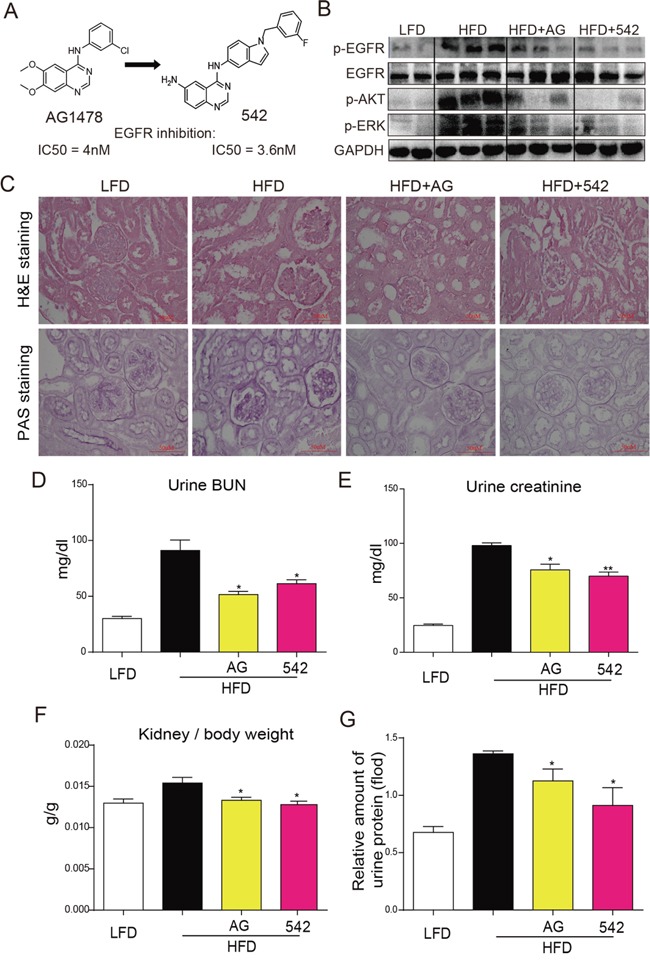
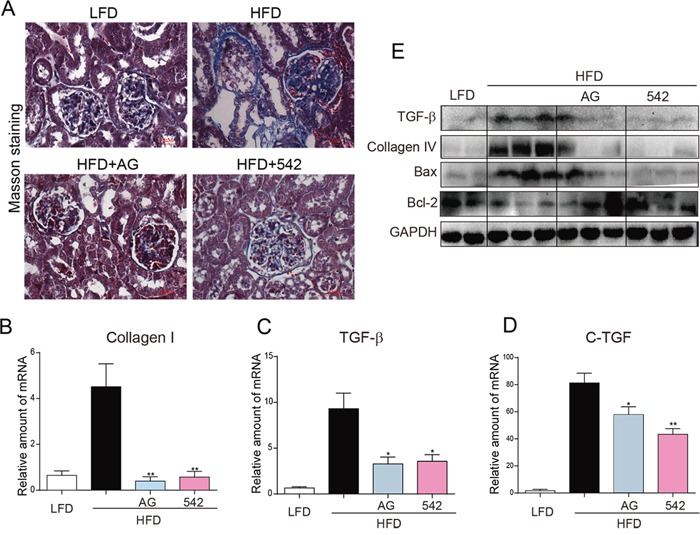
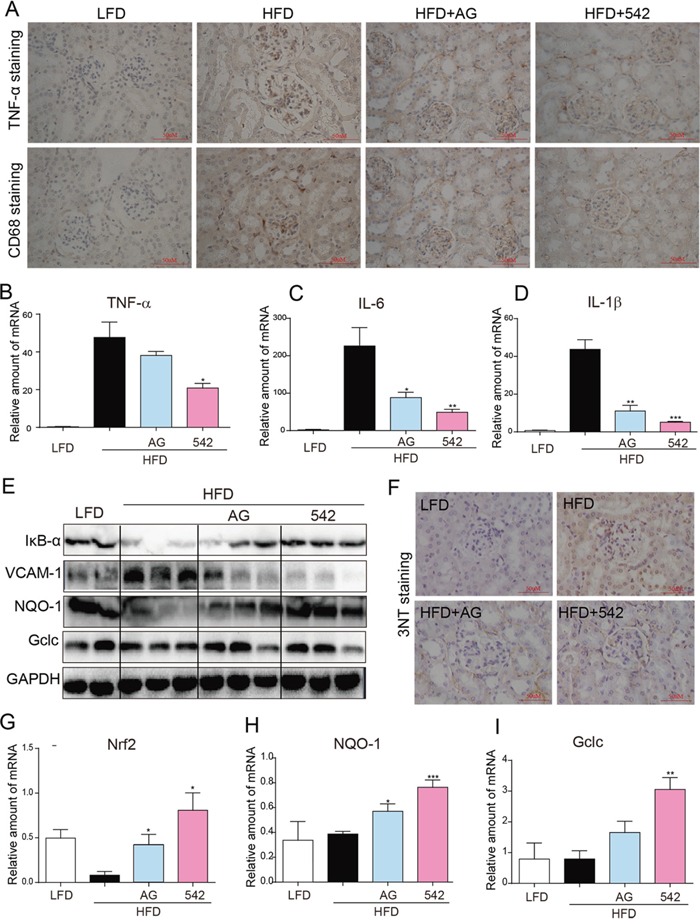
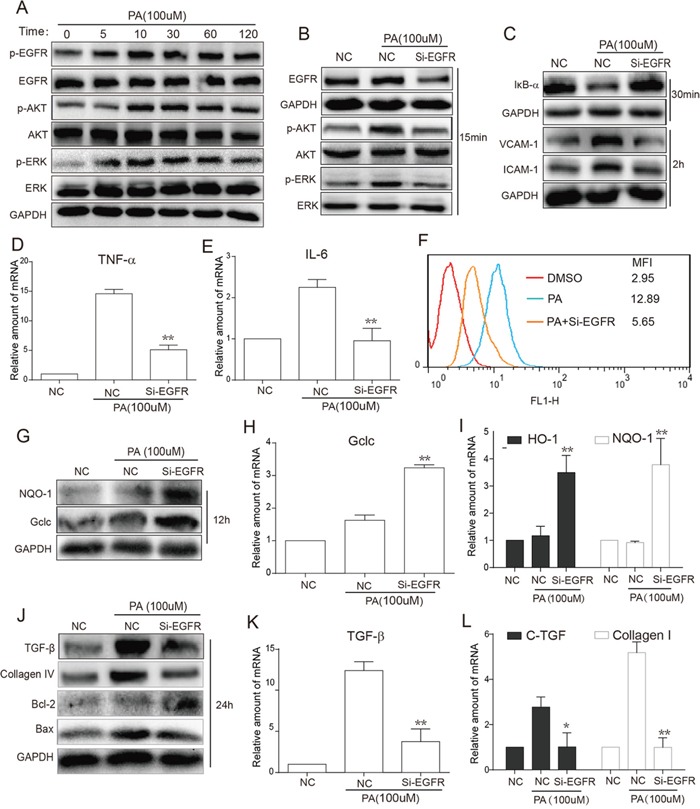
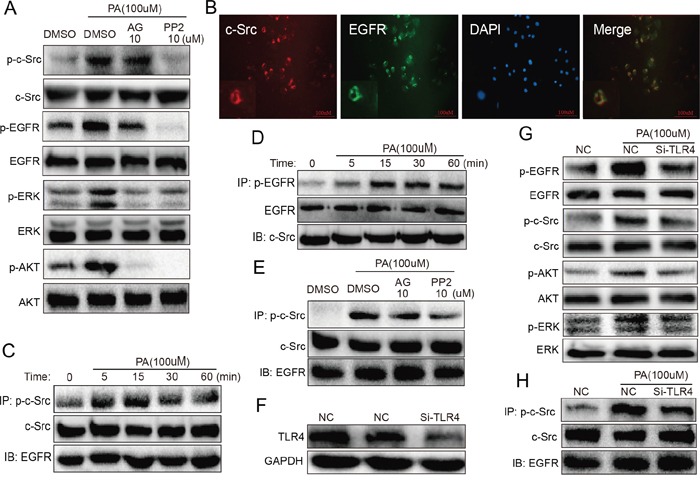
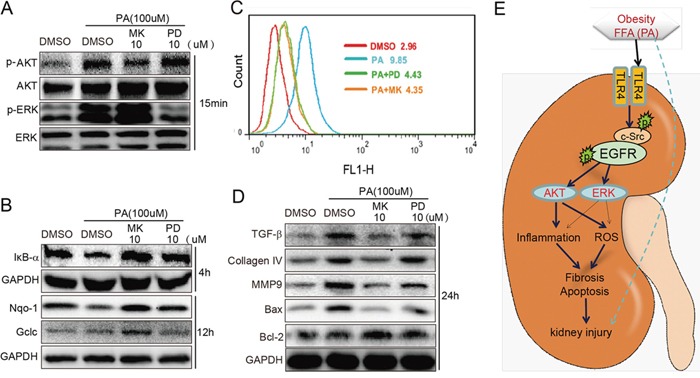
Previous studies have implicated inflammation, oxidative stress, and fibrosis as key factors in the development of obesity-induced kidney diseases. Epidermal growth factor receptor (EGFR) plays an important role in cancer development. Recently, the EGFR pathway has been increasingly implicated in chronic cardiovascular diseases via regulating inflammation and oxidative stress. However, it is unclear if EGFR is involved in obesity-related kidney injury. Using ApoE-/- and C57BL/6 mice models and two specific EGFR inhibitors, we investigated the potential effects of EGFR inhibition in the treatment of obesity-related nephropathy and found that EGFR inhibition alleviates renal inflammation, oxidative stress and fibrosis. In NRK-52E cells, we also elucidated the mechanism behind hyperlipidemia-induced EGFR activation. We observed that c-Src and EGFR forms a complex, and following PA stimulation, it is the successive phosphorylation, not formation, of the c-Src/EGFR complex that results in the subsequent cascade activation. Second, we found that TLR4 regulates the activation EGFR pathway mainly through the phosphorylation of the c-Src/EGFR complex. These results demonstrate the detrimental role of EGFR in the pathogenesis of obesity-related nephropathy, provide a new understanding of the mechanism behind hyperlipidemia/FFA-induced EGFR activation, and support the use of EGFR inhibitors in the treatment of obesity-induced kidney diseases.
Keywords: c-Src; epidermal growth factor receptor; obesity; obesity-induced kidney injury; palmitate.
Conflict of interest statement
All the authors declare no competing financial interest.
Figures
Similar articles
-
EGFR Inhibition Blocks Palmitic Acid-induced inflammation in cardiomyocytes and Prevents Hyperlipidemia-induced Cardiac Injury in Mice.Sci Rep. 2016 Apr 18;6:24580. doi: 10.1038/srep24580. Sci Rep. 2016. PMID: 27087279 Free PMC article.
-
New EGFR inhibitor, 453, prevents renal fibrosis in angiotensin II-stimulated mice.Eur J Pharmacol. 2016 Oct 15;789:421-430. doi: 10.1016/j.ejphar.2016.08.009. Epub 2016 Aug 4. Eur J Pharmacol. 2016. PMID: 27497883
-
Inhibition of epidermal growth factor receptor attenuates LPS-induced inflammation and acute lung injury in rats.Oncotarget. 2017 Apr 18;8(16):26648-26661. doi: 10.18632/oncotarget.15790. Oncotarget. 2017. PMID: 28460454 Free PMC article.
-
Role of Epidermal Growth Factor Receptor (EGFR) and Its Ligands in Kidney Inflammation and Damage.Mediators Inflamm. 2018 Dec 23;2018:8739473. doi: 10.1155/2018/8739473. eCollection 2018. Mediators Inflamm. 2018. PMID: 30670929 Free PMC article. Review.
-
The epidermal growth factor receptor pathway in chronic kidney diseases.Nat Rev Nephrol. 2016 Aug;12(8):496-506. doi: 10.1038/nrneph.2016.91. Epub 2016 Jul 4. Nat Rev Nephrol. 2016. PMID: 27374915 Review.
Cited by
-
Src family kinases in chronic kidney disease.Am J Physiol Renal Physiol. 2017 Sep 1;313(3):F721-F728. doi: 10.1152/ajprenal.00141.2017. Epub 2017 Jun 14. Am J Physiol Renal Physiol. 2017. PMID: 28615246 Free PMC article. Review.
-
The Blockade of TACE-Dependent EGF Receptor Activation by Losartan-Erlotinib Combination Attenuates Renal Fibrosis Formation in 5/6-Nephrectomized Rats Under Vitamin D Deficiency.Front Med (Lausanne). 2021 Jan 5;7:609158. doi: 10.3389/fmed.2020.609158. eCollection 2020. Front Med (Lausanne). 2021. PMID: 33469545 Free PMC article.
-
Investigating the Underlying Mechanisms of Ardisia japonica Extract's Anti-Blood-Stasis Effect via Metabolomics and Network Pharmacology.Molecules. 2023 Oct 27;28(21):7301. doi: 10.3390/molecules28217301. Molecules. 2023. PMID: 37959722 Free PMC article.
-
Impact of Di-(2-Ethylhexyl)-Phthalate on Metabolic Syndrome: Insights from Network Toxicology and Molecular Docking and Dynamics.Diabetes Metab Syndr Obes. 2025 Jul 8;18:2277-2288. doi: 10.2147/DMSO.S523668. eCollection 2025. Diabetes Metab Syndr Obes. 2025. PMID: 40655169 Free PMC article.
-
Resistin facilitates metastasis of lung adenocarcinoma through the TLR4/Src/EGFR/PI3K/NF-κB pathway.Cancer Sci. 2018 Aug;109(8):2391-2400. doi: 10.1111/cas.13704. Epub 2018 Jul 20. Cancer Sci. 2018. PMID: 29927028 Free PMC article.
References
-
- Lafuente MP, Villegas-Perez MP, Selles-Navarro I, Mayor-Torroglosa S, Miralles de Imperial J, Vidal-Sanz M. Retinal ganglion cell death after acute retinal ischemia is an ongoing process whose severity and duration depends on the duration of the insult. Neuroscience. 2002;109:157–168. - PubMed
-
- Bianchi ME. DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol. 2007;81:1–5. - PubMed
-
- Vogl T, Tenbrock K, Ludwig S, Leukert N, Ehrhardt C, van Zoelen MA, Nacken W, Foell D, van der Poll T, Sorg C, Roth J. Mrp8 and Mrp14 are endogenous activators of Toll-like receptor 4, promoting lethal, endotoxin-induced shock. Nat Med. 2007;13:1042–1049. - PubMed
-
- Kondo Y, Ikeda K, Tokuda N, Nishitani C, Ohto U, Akashi-Takamura S, Ito Y, Uchikawa M, Kuroki Y, Taguchi R, Miyake K, Zhang Q, Furukawa K, Furukawa K. TLR4-MD-2 complex is negatively regulated by an endogenous ligand, globotetraosylceramide. Proc Natl Acad Sci U S A. 2013;110:4714–4719. - PMC - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
