

Oxidative stress at low levels can induce clustered DNA lesions leading to NHEJ mediated mutations
- PMID: 27015367
- PMCID: PMC5041911
- DOI: 10.18632/oncotarget.8298
Oxidative stress at low levels can induce clustered DNA lesions leading to NHEJ mediated mutations
Abstract
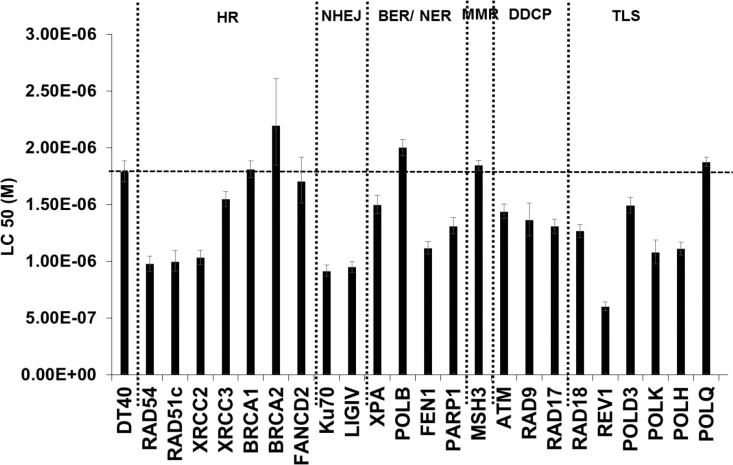
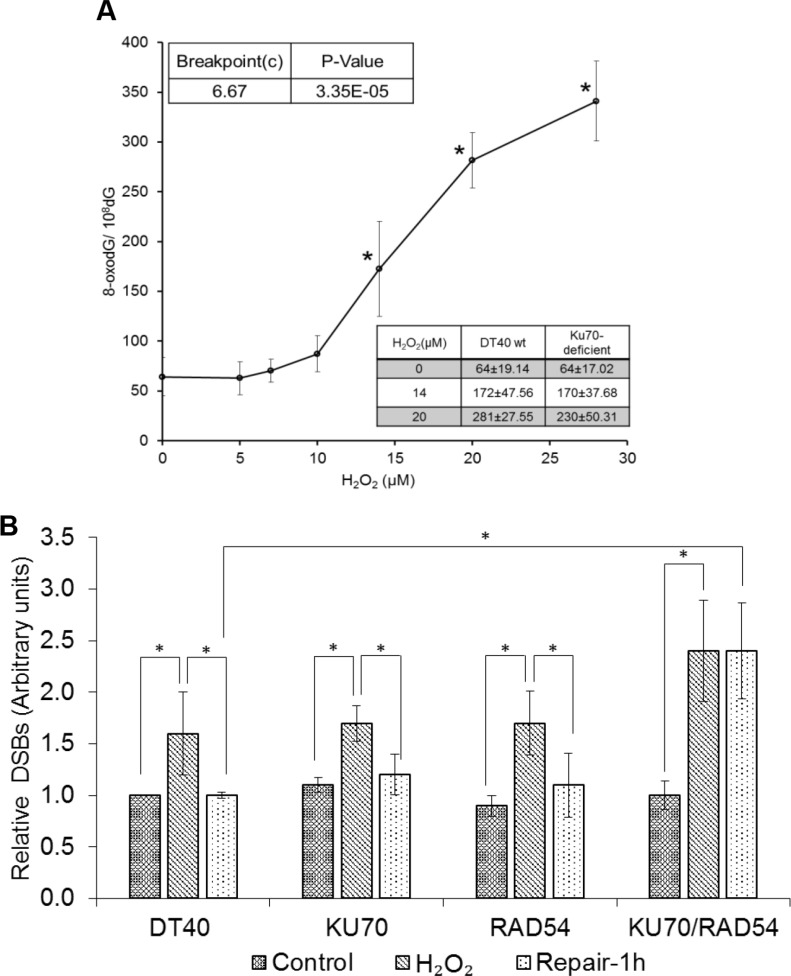
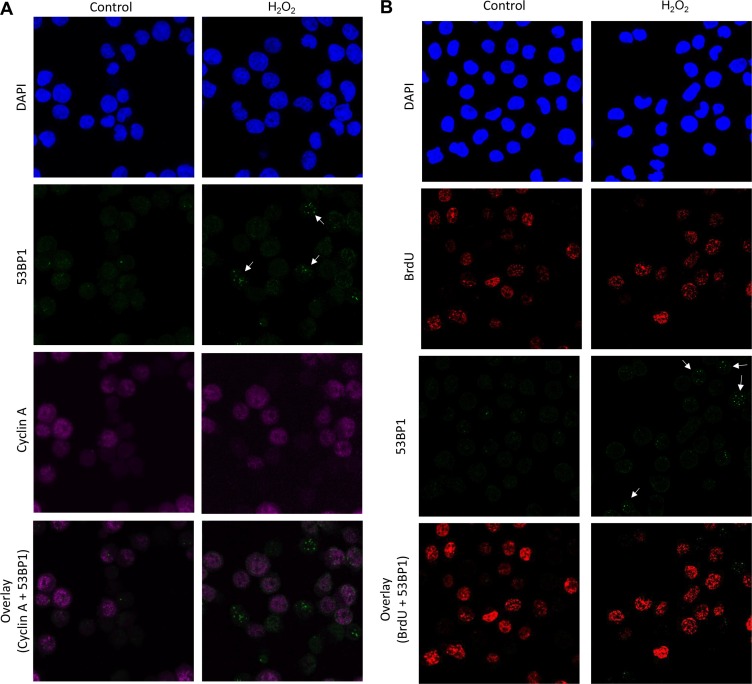
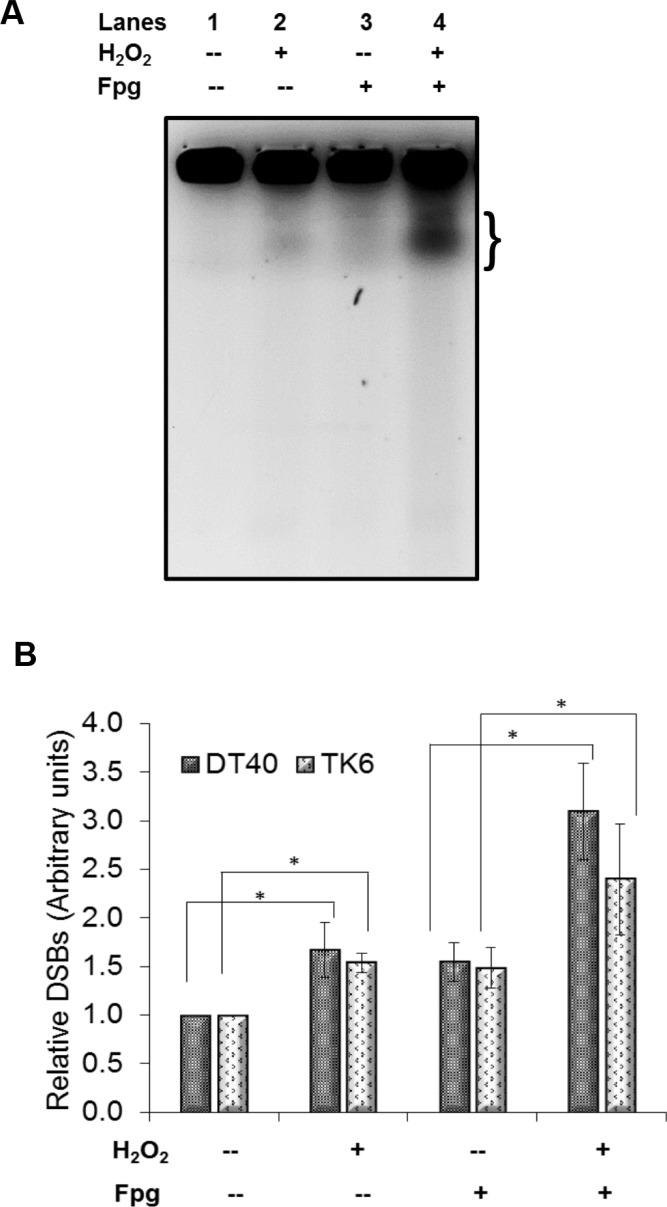
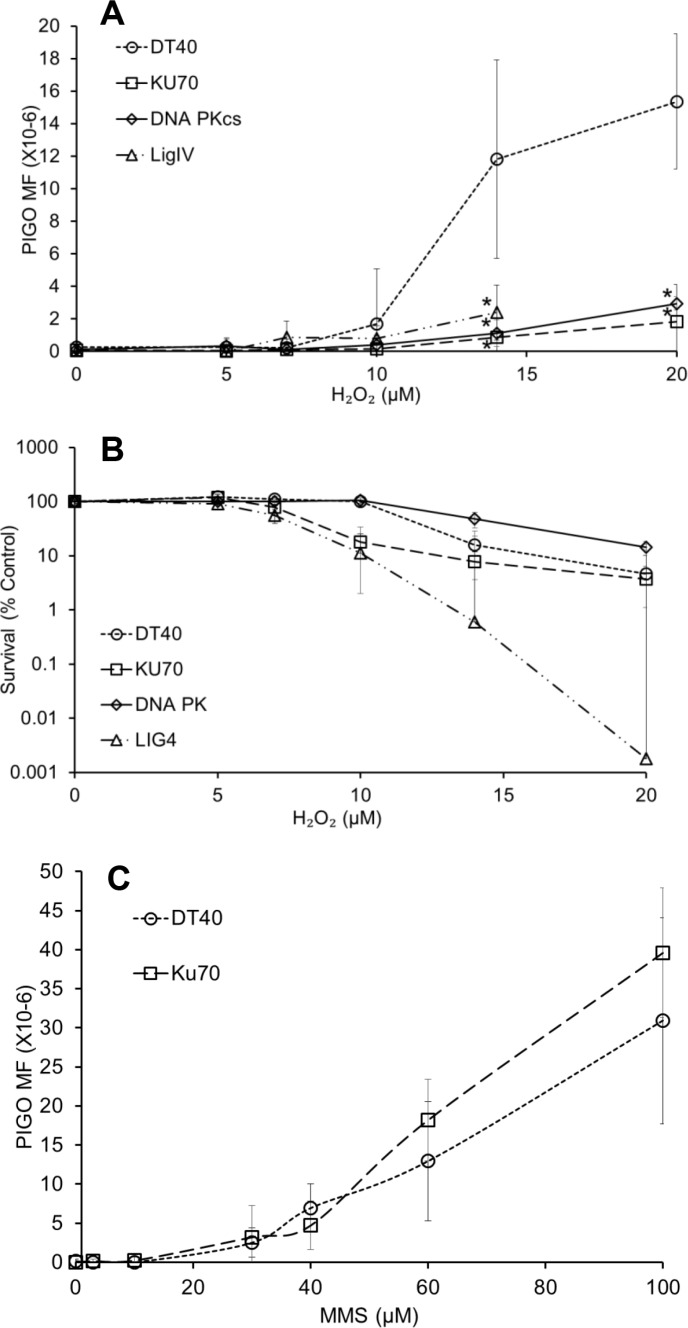
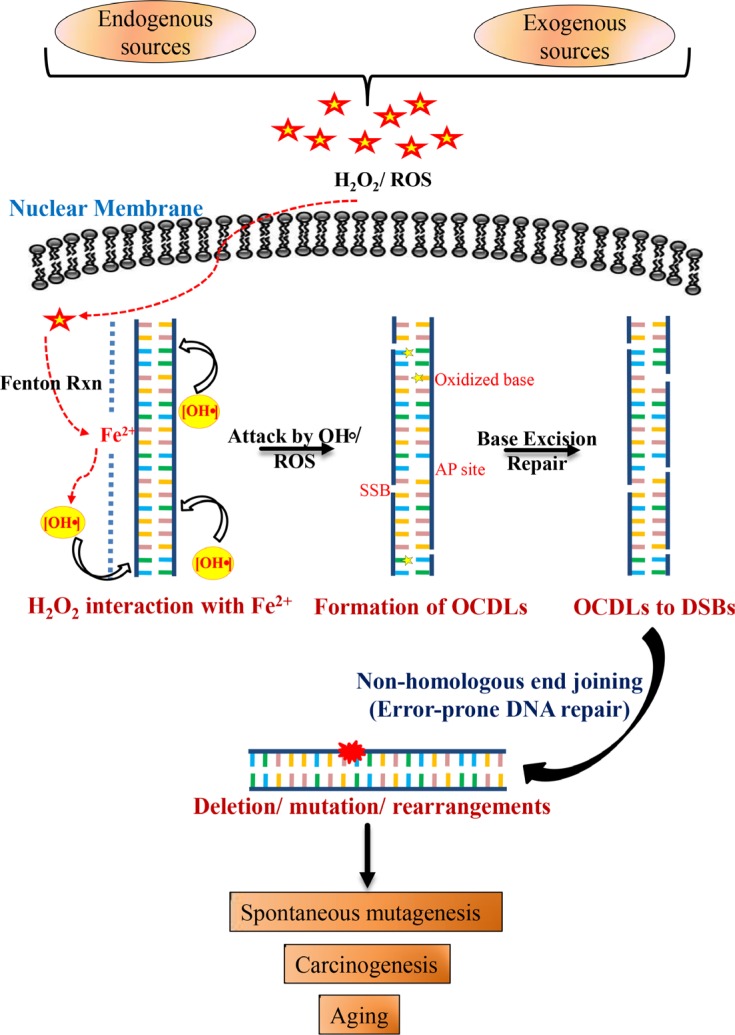
DNA damage and mutations induced by oxidative stress are associated with various different human pathologies including cancer. The facts that most human tumors are characterized by large genome rearrangements and glutathione depletion in mice results in deletions in DNA suggest that reactive oxygen species (ROS) may cause gene and chromosome mutations through DNA double strand breaks (DSBs). However, the generation of DSBs at low levels of ROS is still controversial. In the present study, we show that H2O2 at biologically-relevant levels causes a marked increase in oxidative clustered DNA lesions (OCDLs) with a significant elevation of replication-independent DSBs. Although it is frequently reported that OCDLs are fingerprint of high-energy IR, our results indicate for the first time that H2O2, even at low levels, can also cause OCDLs leading to DSBs specifically in G1 cells. Furthermore, a reverse genetic approach revealed a significant contribution of the non-homologous end joining (NHEJ) pathway in H2O2-induced DNA repair & mutagenesis. This genomic instability induced by low levels of ROS may be involved in spontaneous mutagenesis and the etiology of a wide variety of human diseases like chronic inflammation-related disorders, carcinogenesis, neuro-degeneration and aging.
Keywords: NHEJ; clustered DNA lesions; double strand breaks; mutations; oxidative stress.
Conflict of interest statement
Conflicts of Interest: None declared.
Figures
References
-
- Evans MD, Dizdaroglu M, Cooke MS. Oxidative DNA damage and disease: induction, repair and significance. Mutat Res. 2004;567:1–61. - PubMed
-
- Toyokuni S, Okamoto K, Yodoi J, Hiai H. Persistent oxidative stress in cancer. FEBS Lett. 1995;358:1–3. - PubMed
-
- Lieber MR, Karanjawala ZE. Ageing, repetitive genomes and DNA damage. Nat Rev Mol Cell Biol. 2004;5:69–75. - PubMed
-
- Ames BN. Endogenous DNA damage as related to cancer and aging. Mutat Res. 1989;214:41–46. - PubMed
-
- Guyton KZ, Kensler TW. Oxidative mechanisms in carcinogenesis. Br Med Bull. 1993;49:523–544. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
