

Motor neuron degeneration in spastic paraplegia 11 mimics amyotrophic lateral sclerosis lesions
- PMID: 27016404
- PMCID: PMC5839621
- DOI: 10.1093/brain/aww061
Motor neuron degeneration in spastic paraplegia 11 mimics amyotrophic lateral sclerosis lesions
Abstract
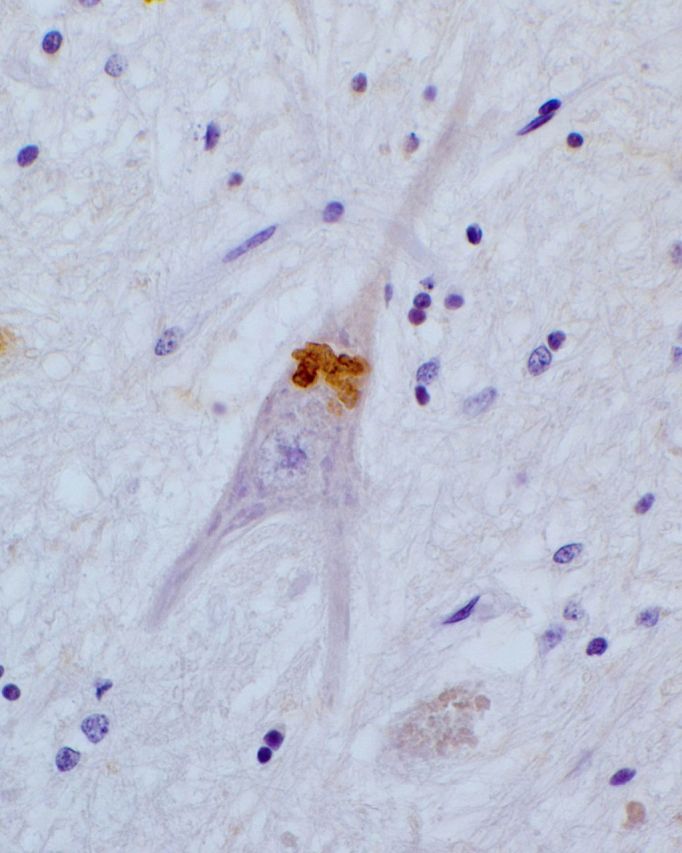
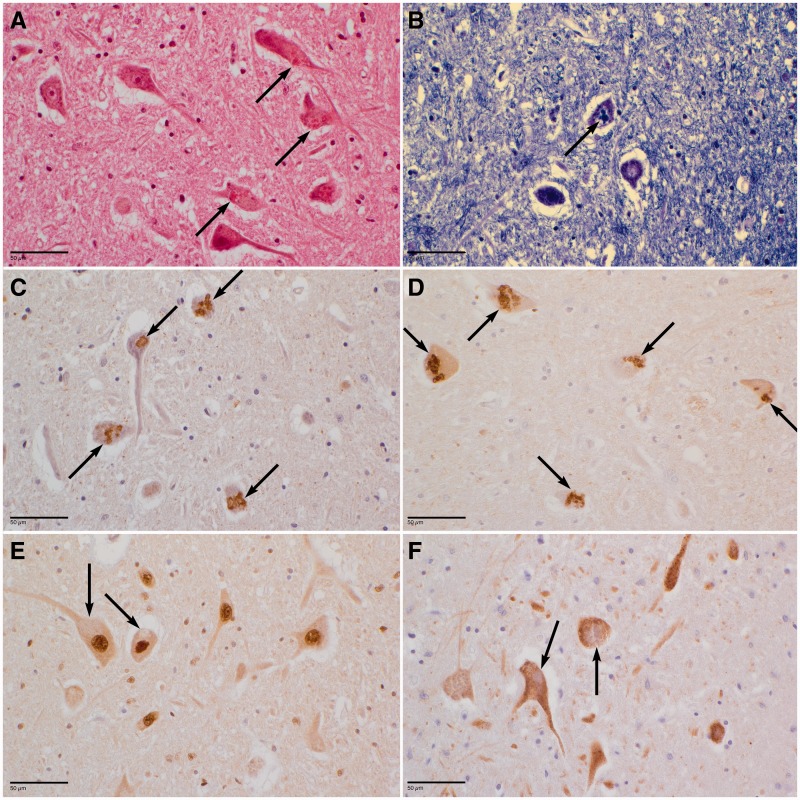
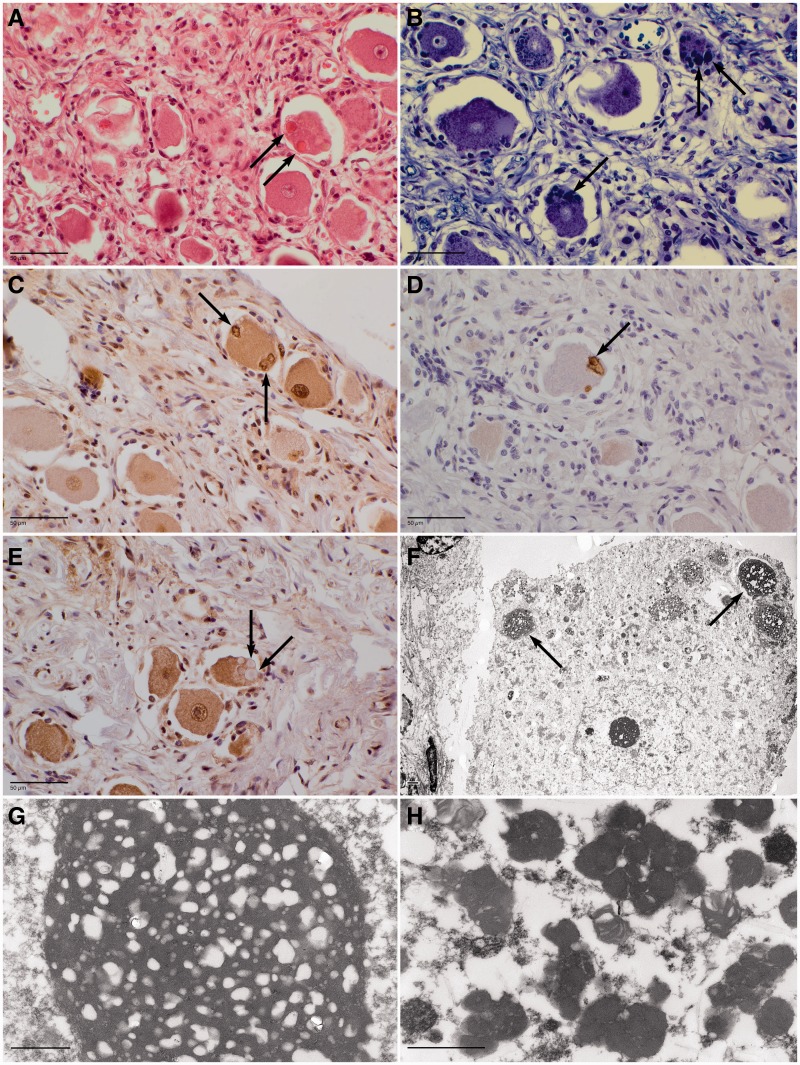
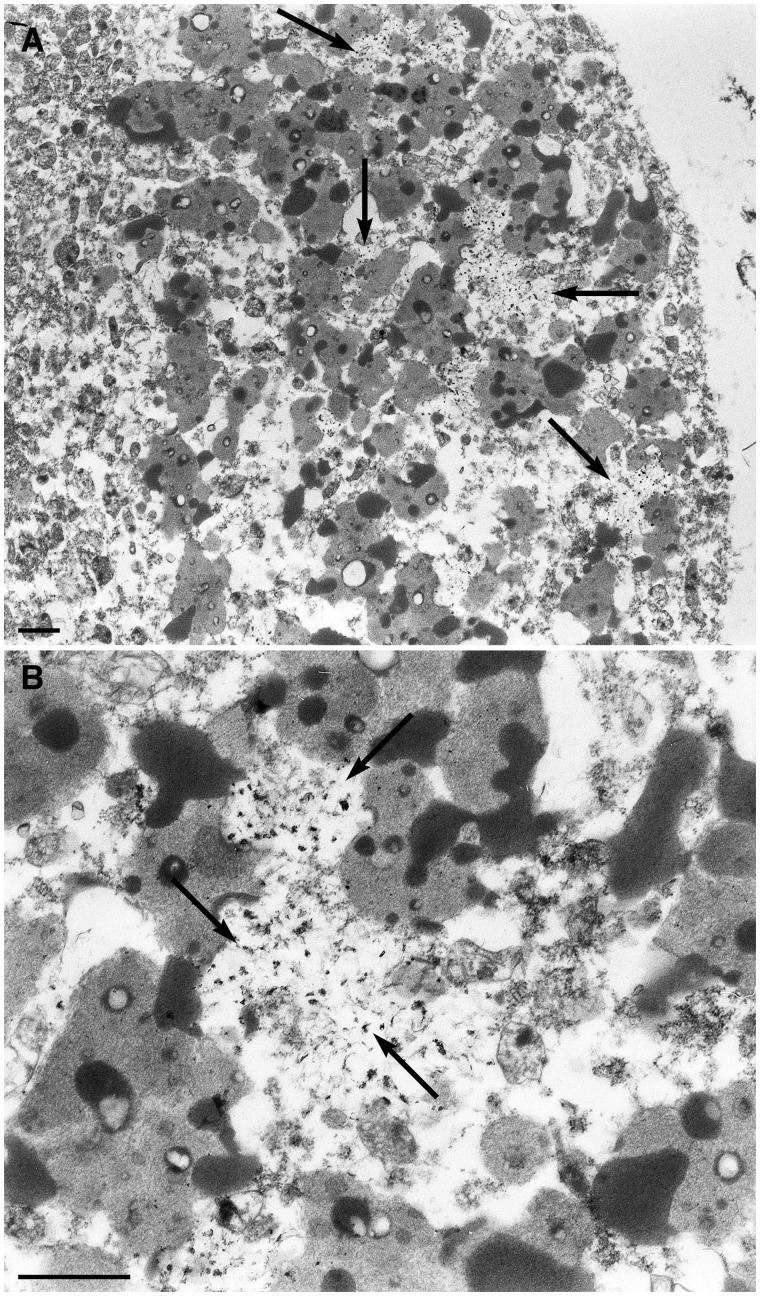
The most common form of autosomal recessive hereditary spastic paraplegia is caused by mutations in the SPG11/KIAA1840 gene on chromosome 15q. The nature of the vast majority of SPG11 mutations found to date suggests a loss-of-function mechanism of the encoded protein, spatacsin. The SPG11 phenotype is, in most cases, characterized by a progressive spasticity with neuropathy, cognitive impairment and a thin corpus callosum on brain MRI. Full neuropathological characterization has not been reported to date despite the description of >100 SPG11 mutations. We describe here the clinical and pathological features observed in two unrelated females, members of genetically ascertained SPG11 families originating from Belgium and Italy, respectively. We confirm the presence of lesions of motor tracts in medulla oblongata and spinal cord associated with other lesions of the central nervous system. Interestingly, we report for the first time pathological hallmarks of SPG11 in neurons that include intracytoplasmic granular lysosome-like structures mainly in supratentorial areas, and others in subtentorial areas that are partially reminiscent of those observed in amyotrophic lateral sclerosis, such as ubiquitin and p62 aggregates, except that they are never labelled with anti-TDP-43 or anti-cystatin C. The neuropathological overlap with amyotrophic lateral sclerosis, associated with some shared clinical manifestations, opens up new fields of investigation in the physiopathological continuum of motor neuron degeneration.
Keywords: amyotrophic lateral sclerosis; lipofuscin; lysosome; spastic paraplegia 11; spatacsin.
© The Author (2016). Published by Oxford University Press on behalf of the Guarantors of Brain. All rights reserved. For Permissions, please email: journals.permissions@oup.com.
Figures
References
-
- Boukhris A, Stevanin G, Feki I, Denora P, Elleuch N, Miladi MI, et al. Tunisian hereditary spastic paraplegias: clinical variability supported by genetic heterogeneity. Clin Genet 2009; 75: 527–36. - PubMed
-
- Coutinho P, Ruano L, Loureiro JL, Cruz VT, Barros J, Tuna A, et al. Hereditary ataxia and spastic paraplegia in Portugal: a population-based prevalence study. JAMA Neurol 2013; 70: 746–55. - PubMed
-
- Crimella C, Arnoldi A, Crippa F, Mostacciuolo ML, Boaretto F, Sironi M, et al. Point mutations and a large intragenic deletion in SPG11 in complicated spastic paraplegia without thin corpus callosum. J Med Genet 2009; 46: 345–51. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
