

Uncovering Adaptation from Sequence Data: Lessons from Genome Resequencing of Four Cattle Breeds
- PMID: 27017625
- PMCID: PMC4858790
- DOI: 10.1534/genetics.115.181594
Uncovering Adaptation from Sequence Data: Lessons from Genome Resequencing of Four Cattle Breeds
Abstract
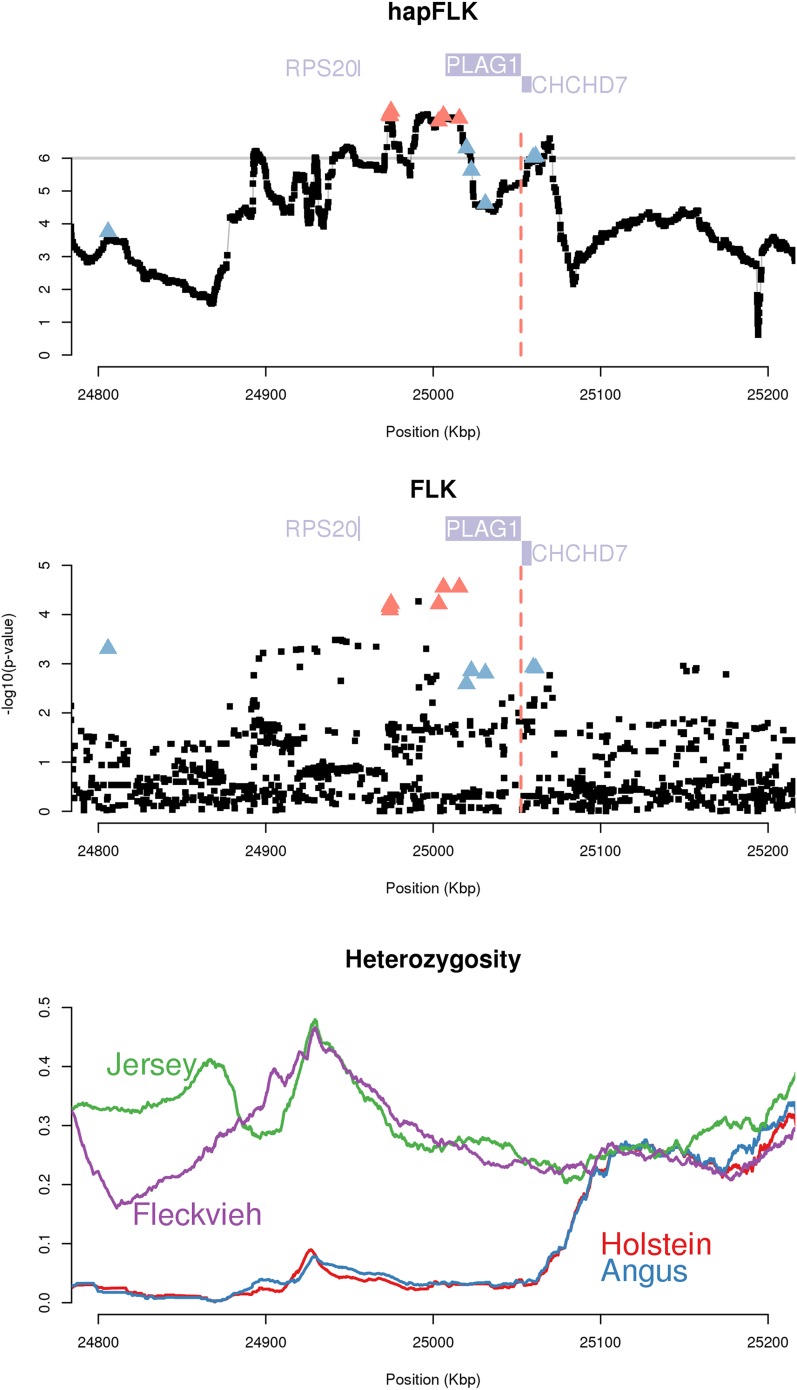
Detecting the molecular basis of adaptation is one of the major questions in population genetics. With the advance in sequencing technologies, nearly complete interrogation of genome-wide polymorphisms in multiple populations is becoming feasible in some species, with the expectation that it will extend quickly to new ones. Here, we investigate the advantages of sequencing for the detection of adaptive loci in multiple populations, exploiting a recently published data set in cattle (Bos taurus). We used two different approaches to detect statistically significant signals of positive selection: a within-population approach aimed at identifying hard selective sweeps and a population-differentiation approach that can capture other selection events such as soft or incomplete sweeps. We show that the two methods are complementary in that they indeed capture different kinds of selection signatures. Our study confirmed some of the well-known adaptive loci in cattle (e.g., MC1R, KIT, GHR, PLAG1, NCAPG/LCORL) and detected some new ones (e.g., ARL15, PRLR, CYP19A1, PPM1L). Compared to genome scans based on medium- or high-density SNP data, we found that sequencing offered an increased detection power and a higher resolution in the localization of selection signatures. In several cases, we could even pinpoint the underlying causal adaptive mutation or at least a very small number of possible candidates (e.g., MC1R, PLAG1). Our results on these candidates suggest that a vast majority of adaptive mutations are likely to be regulatory rather than protein-coding variants.
Keywords: FST; domestication; linkage disequilibrium; next-generation sequencing; selective sweeps.
Copyright © 2016 by the Genetics Society of America.
Figures
References
-
- Bionaz M., Loor J. J., 2008. Acsl1, agpat6, fabp3, lpin1, and slc27a6 are the most abundant isoforms in bovine mammary tissue and their expression is affected by stage of lactation. J. Nutr. 138: 1019–1024. - PubMed
-
- Biswas S., Akey J. M., 2006. Genomic insights into positive selection. Trends Genet. 22: 437–446. - PubMed
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
