

Alternative Roles of STAT3 and MAPK Signaling Pathways in the MMPs Activation and Progression of Lung Injury Induced by Cigarette Smoke Exposure in ACE2 Knockout Mice
- PMID: 27019629
- PMCID: PMC4807164
- DOI: 10.7150/ijbs.13379
Alternative Roles of STAT3 and MAPK Signaling Pathways in the MMPs Activation and Progression of Lung Injury Induced by Cigarette Smoke Exposure in ACE2 Knockout Mice
Abstract
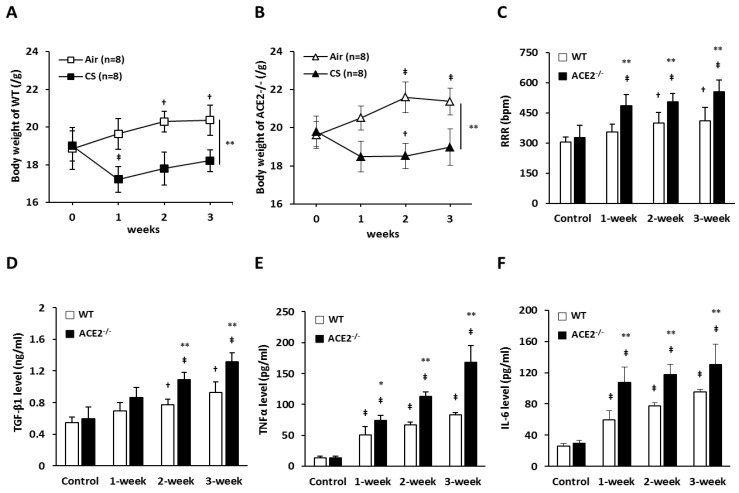
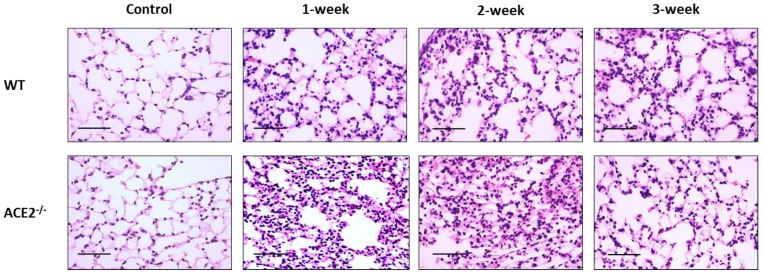
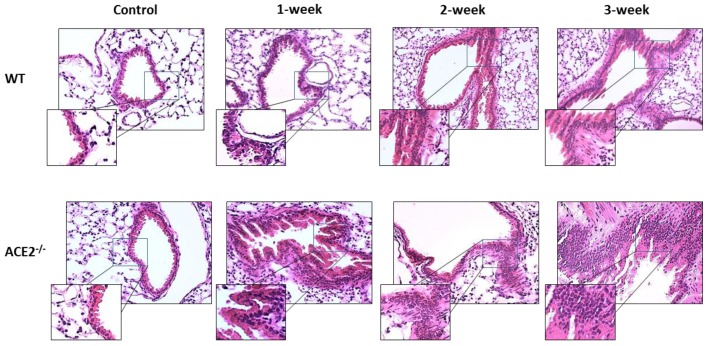
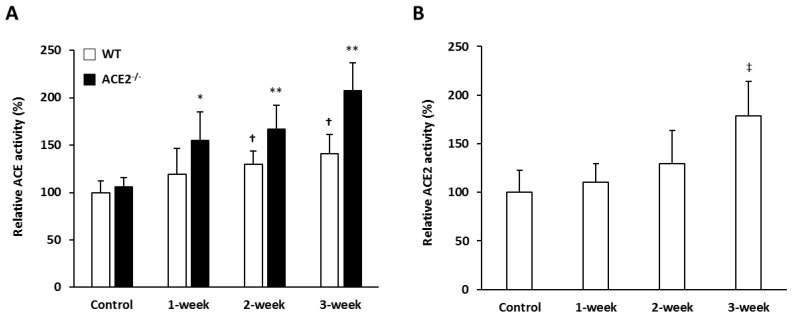
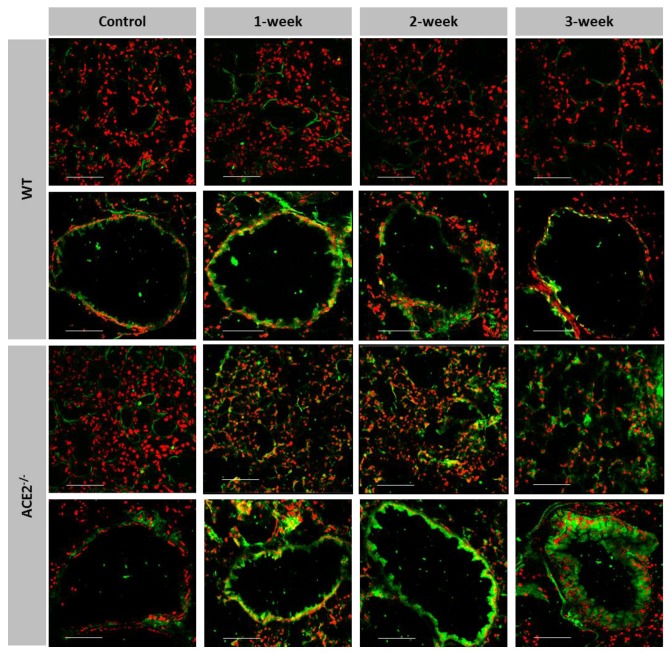
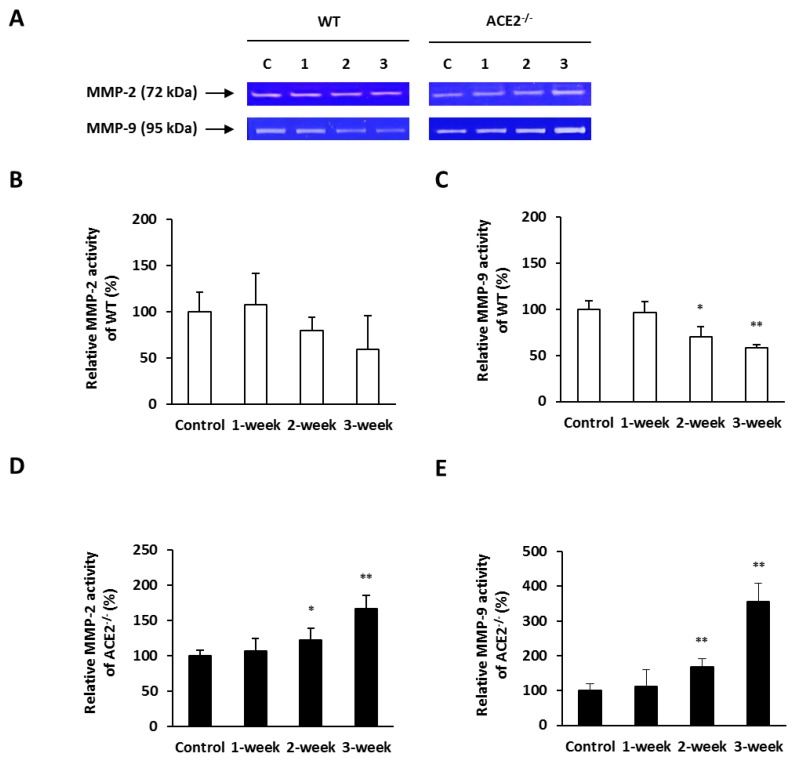
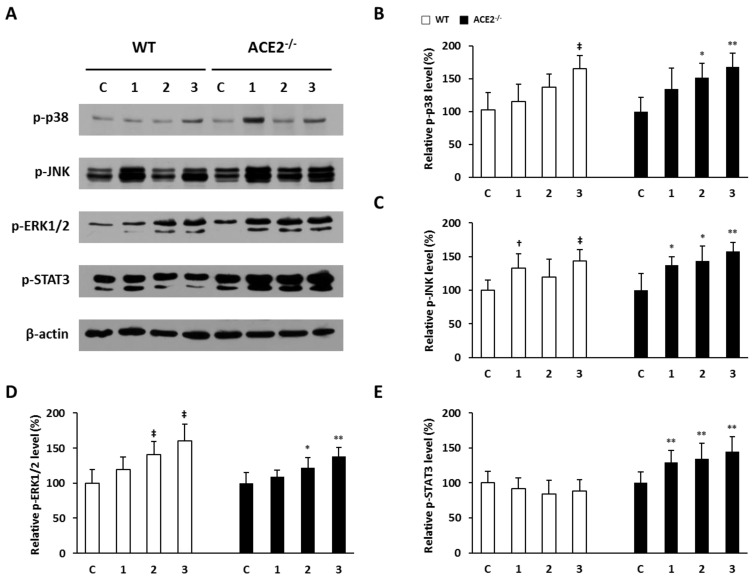
Inflammation-mediated abnormalities in the renin-angiotensin system (RAS) and expression of matrix metalloproteinases (MMPs) are implicated in the pathogenesis of lung injury. Angiotensin converting enzyme II (ACE2), an angiotensin converting enzyme (ACE) homologue that displays antagonist effects on ACE/angiotensin II (Ang II) axis, could also play a protective role against lung diseases. However, the relationship between ACE2 and MMPs activation in lung injury is still largely unclear. The purpose of this study is to investigate whether MMPs activity could be affected by ACE2 and which ACE2 derived signaling pathways could be also involved via using a mouse model with lung injury induced by cigarette smoke (CS) exposure for 1 to 3 weeks. Wild-type (WT; C57BL/6) and ACE2 KO mice (ACE2(-/-)) were utilized to study CS-induced lung injury. Increases in the resting respiratory rate (RRR), pulmonary immunokines, leukocyte infiltration and bronchial hyperplasia were observed in the CS-exposed mice. Compared to WT mice, more serious physiopathological changes were found in ACE2(-/-) mice in the first week of CS exposure. CS exposure increased pulmonary ACE and ACE2 activities in WT mice, and significantly increased ACE in ACE2(-/-) mice. Furthermore, the activity of pulmonary MMPs was decreased in CS-exposed WT mice, whereas this activity was increased in ACE2(-/-) mice. CS exposure increased the pulmonary p-p38, p-JNK and p-ERK1/2 level in all mice. In ACE2(-/-) mice, a significant increase p-STAT3 signaling was detected; however, no effect was observed on the p-STAT3 level in WT mice. Our results support the hypothesis that ACE2 deficiency influences MMPs activation and STAT3 phosphorylation signaling to promote more pulmonary inflammation in the development of lung injury.
Keywords: angiotensin converting enzyme II; cigarette smoke; lung injury; matrix metalloproteinases; renin-angiotensin system.
Conflict of interest statement
Competing Interests: The authors have declared that no competing interest exists.
Figures
References
-
- Rubenfeld GD, Caldwell E, Peabody E. et al. Incidence and outcomes of acute lung injury. N Engl J Med. 2005;353:1685–93. - PubMed
-
- Wright JL, Zhou S, Churg A. Pulmonary hypertension and vascular oxidative damage in cigarette smoke exposed eNOS(-/-) mice and human smokers. Inhal Toxicol. 2012;24:732–40. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
