

The Impact of DNA Methylation in Hematopoietic Malignancies
- PMID: 27019871
- PMCID: PMC4806338
- DOI: 10.1016/j.trecan.2015.12.006
The Impact of DNA Methylation in Hematopoietic Malignancies
Abstract
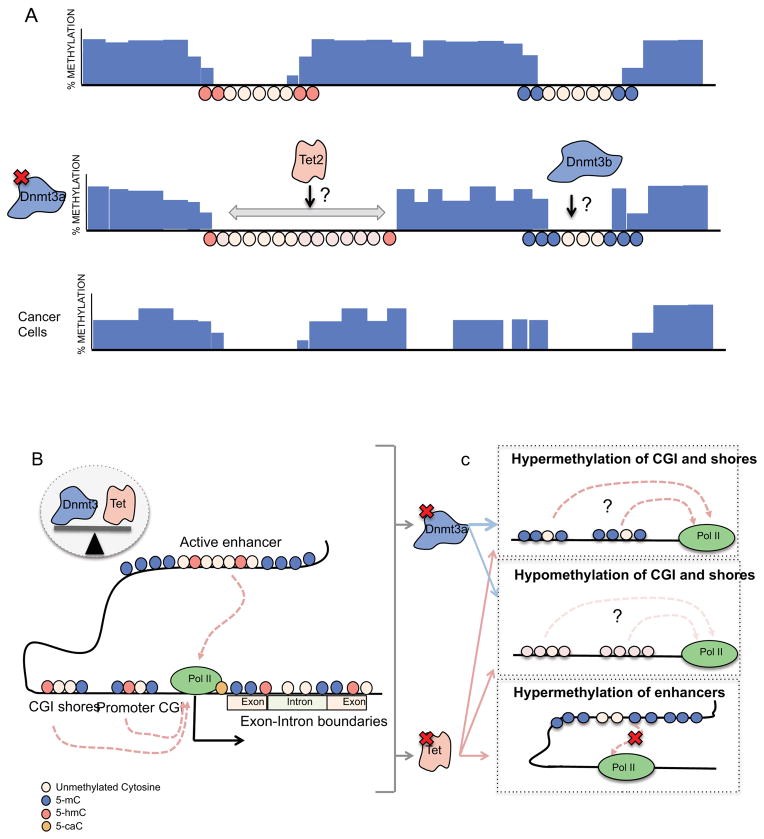
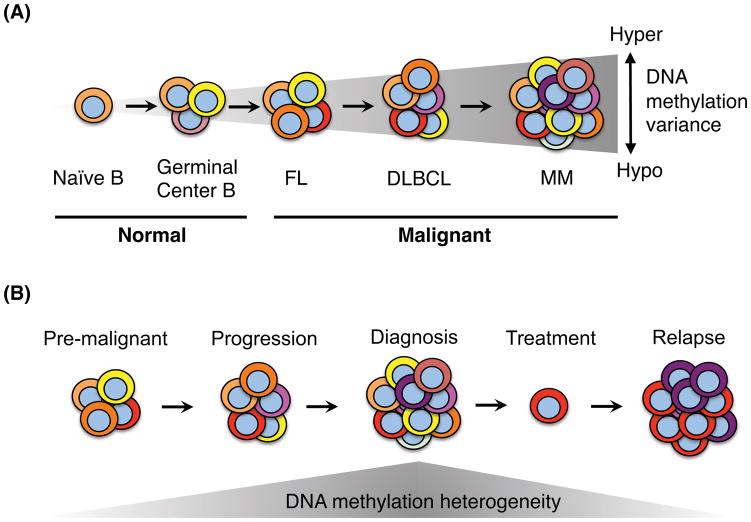
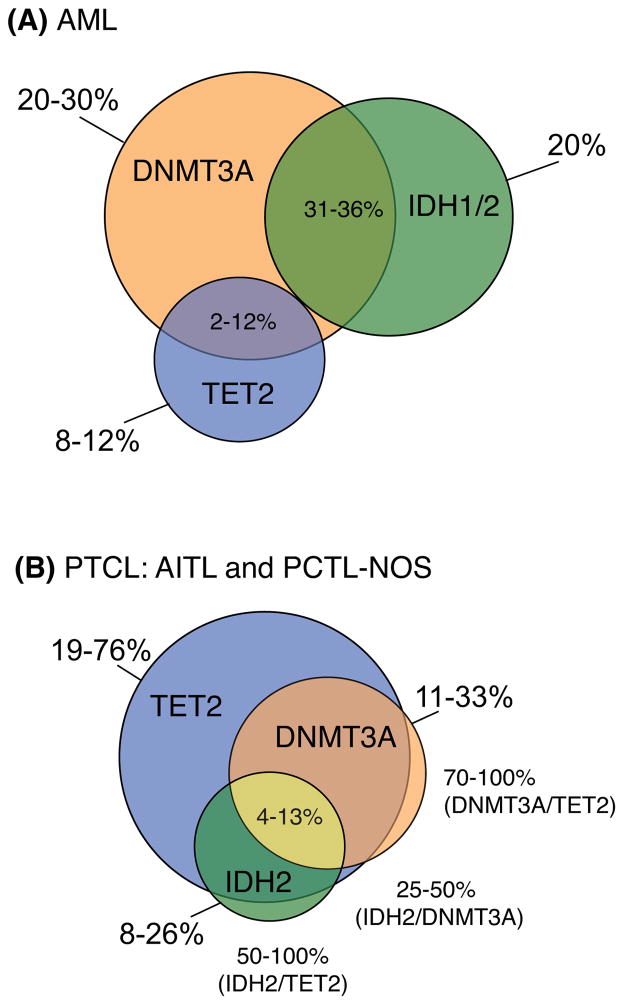
Aberrant DNA methylation is a characteristic feature of cancer including blood malignancies. Mutations in the DNA methylation regulators DNMT3A, TET1/2 and IDH1/2 are recurrent in leukemia and lymphoma. Specific and distinct DNA methylation patterns characterize subtypes of AML and lymphoma. Regulatory regions such as promoter CpG islands, CpG shores and enhancers show changes in methylation during transformation. However, the reported poor correlation between changes in methylation and gene expression in many mouse models and human studies reflects the complexity in the precise molecular mechanism for why aberrant DNA methylation promotes malignancies. This review will summarize current concepts regarding the mechanisms behind aberrant DNA methylation in hematopoietic malignancy and discuss its importance in cancer prognosis, tumor heterogeneity and relapse.
Keywords: DNA methylation; epigenetics; leukemia; lymphoma.
Figures
References
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous