

Genome of Leptomonas pyrrhocoris: a high-quality reference for monoxenous trypanosomatids and new insights into evolution of Leishmania
- PMID: 27021793
- PMCID: PMC4810370
- DOI: 10.1038/srep23704
Genome of Leptomonas pyrrhocoris: a high-quality reference for monoxenous trypanosomatids and new insights into evolution of Leishmania
Abstract
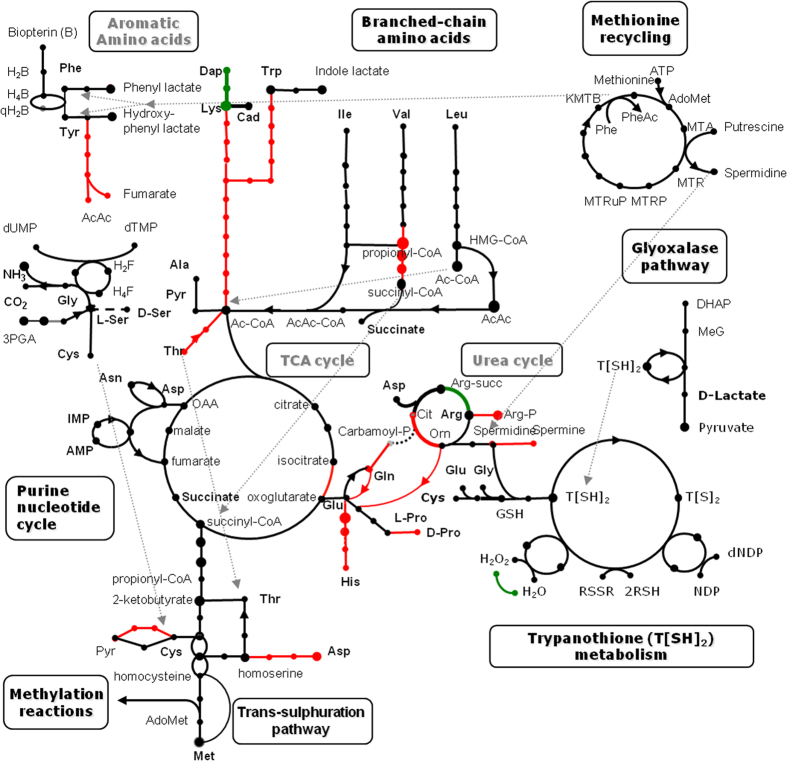
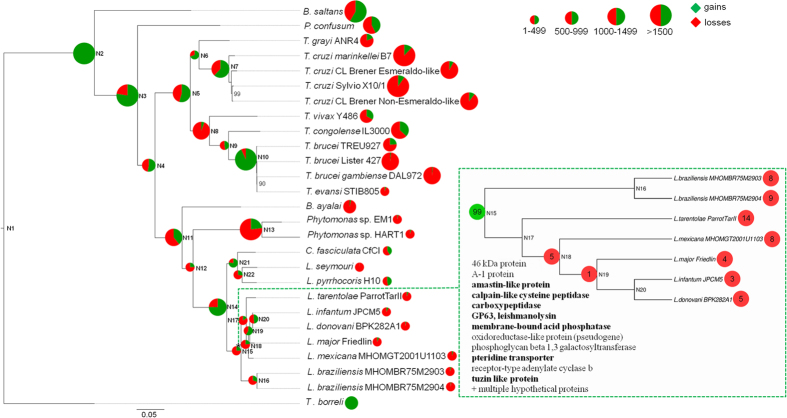
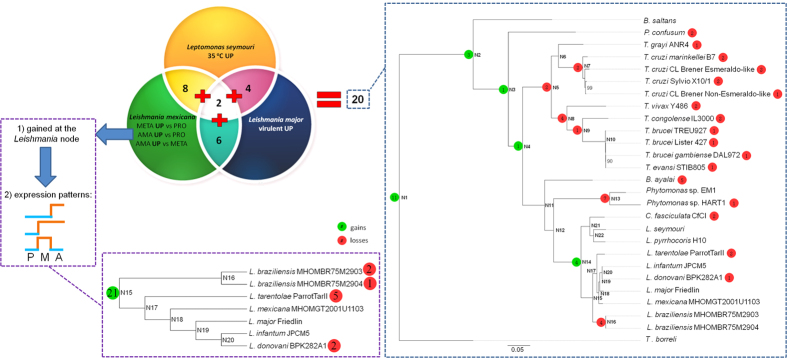
Many high-quality genomes are available for dixenous (two hosts) trypanosomatid species of the genera Trypanosoma, Leishmania, and Phytomonas, but only fragmentary information is available for monoxenous (single-host) trypanosomatids. In trypanosomatids, monoxeny is ancestral to dixeny, thus it is anticipated that the genome sequences of the key monoxenous parasites will be instrumental for both understanding the origin of parasitism and the evolution of dixeny. Here, we present a high-quality genome for Leptomonas pyrrhocoris, which is closely related to the dixenous genus Leishmania. The L. pyrrhocoris genome (30.4 Mbp in 60 scaffolds) encodes 10,148 genes. Using the L. pyrrhocoris genome, we pinpointed genes gained in Leishmania. Among those genes, 20 genes with unknown function had expression patterns in the Leishmania mexicana life cycle suggesting their involvement in virulence. By combining differential expression data for L. mexicana, L. major and Leptomonas seymouri, we have identified several additional proteins potentially involved in virulence, including SpoU methylase and U3 small nucleolar ribonucleoprotein IMP3. The population genetics of L. pyrrhocoris was also addressed by sequencing thirteen strains of different geographic origin, allowing the identification of 1,318 genes under positive selection. This set of genes was significantly enriched in components of the cytoskeleton and the flagellum.
Figures
References
-
- Lukeš J., Skalický T., Týč J., Votýpka J. & Yurchenko V. Evolution of parasitism in kinetoplastid flagellates. Mol Biochem Parasitol 195, 115–122 (2014). - PubMed
-
- Maslov D. A., Votýpka J., Yurchenko V. & Lukeš J. Diversity and phylogeny of insect trypanosomatids: all that is hidden shall be revealed. Trends Parasitol 29, 43–52 (2013). - PubMed
-
- Votýpka J. et al. Cosmopolitan distribution of a trypanosomatid Leptomonas pyrrhocoris. Protist 163, 616–631 (2012). - PubMed
-
- Jirků M., Yurchenko V. Y., Lukeš J. & Maslov D. A. New species of insect trypanosomatids from Costa Rica and the proposal for a new subfamily within the Trypanosomatidae. J Eukaryot Microbiol. 59, 537–547 (2012). - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
