

How many biological replicates are needed in an RNA-seq experiment and which differential expression tool should you use?
- PMID: 27022035
- PMCID: PMC4878611
- DOI: 10.1261/rna.053959.115
How many biological replicates are needed in an RNA-seq experiment and which differential expression tool should you use?
Erratum in
-
Erratum: How many biological replicates are needed in an RNA-seq experiment and which differential expression tool should you use?RNA. 2016 Oct;22(10):1641. doi: 10.1261/rna.058339.116. RNA. 2016. PMID: 27638913 Free PMC article. No abstract available.
Abstract
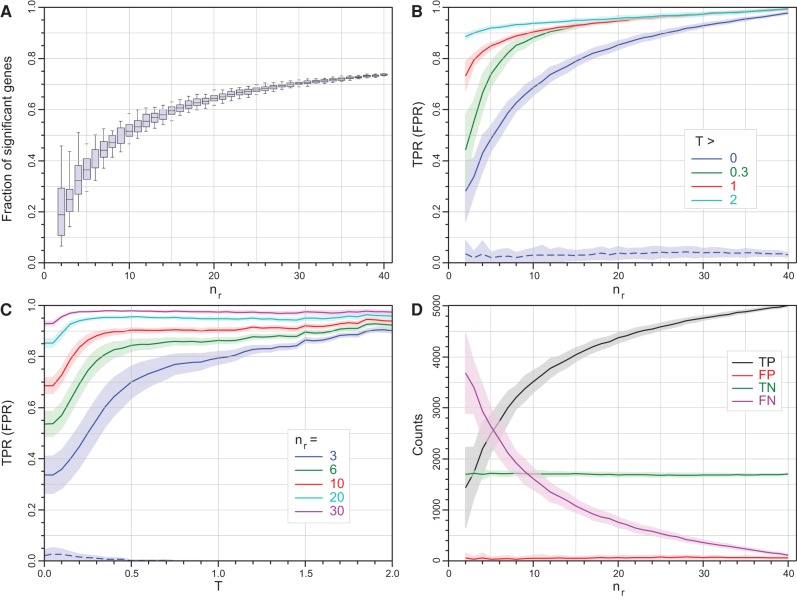
RNA-seq is now the technology of choice for genome-wide differential gene expression experiments, but it is not clear how many biological replicates are needed to ensure valid biological interpretation of the results or which statistical tools are best for analyzing the data. An RNA-seq experiment with 48 biological replicates in each of two conditions was performed to answer these questions and provide guidelines for experimental design. With three biological replicates, nine of the 11 tools evaluated found only 20%-40% of the significantly differentially expressed (SDE) genes identified with the full set of 42 clean replicates. This rises to >85% for the subset of SDE genes changing in expression by more than fourfold. To achieve >85% for all SDE genes regardless of fold change requires more than 20 biological replicates. The same nine tools successfully control their false discovery rate at ≲5% for all numbers of replicates, while the remaining two tools fail to control their FDR adequately, particularly for low numbers of replicates. For future RNA-seq experiments, these results suggest that at least six biological replicates should be used, rising to at least 12 when it is important to identify SDE genes for all fold changes. If fewer than 12 replicates are used, a superior combination of true positive and false positive performances makes edgeR and DESeq2 the leading tools. For higher replicate numbers, minimizing false positives is more important and DESeq marginally outperforms the other tools.
Keywords: RNA-seq; benchmarking; differential expression; experimental design; replication; statistical power; yeast.
© 2016 Schurch et al.; Published by Cold Spring Harbor Laboratory Press for the RNA Society.
Figures



References
-
- Becker PB, Horz W. 2002. ATP-dependent nucleosome remodeling. Annu Rev Biochem 71: 247–273. - PubMed
-
- Bellera CA, Julien M, Hanley JA. 2010. Normal approximations to the distributions of the Wilcoxon statistics: accurate to what N? Graphical insights. J Stat Educ 18.
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases