

Integrative NMR for biomolecular research
- PMID: 27023095
- PMCID: PMC4861749
- DOI: 10.1007/s10858-016-0029-x
Integrative NMR for biomolecular research
Abstract
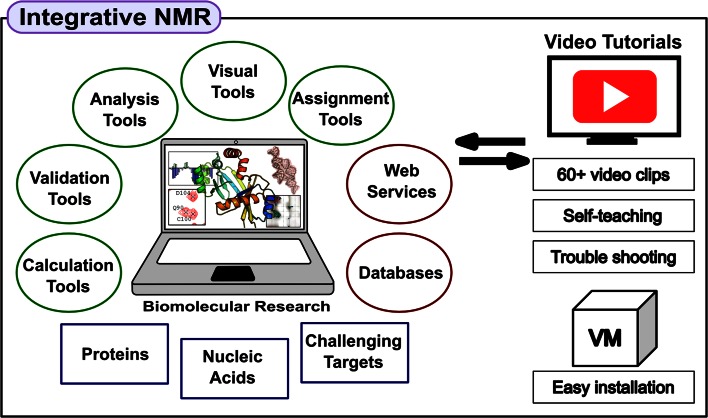
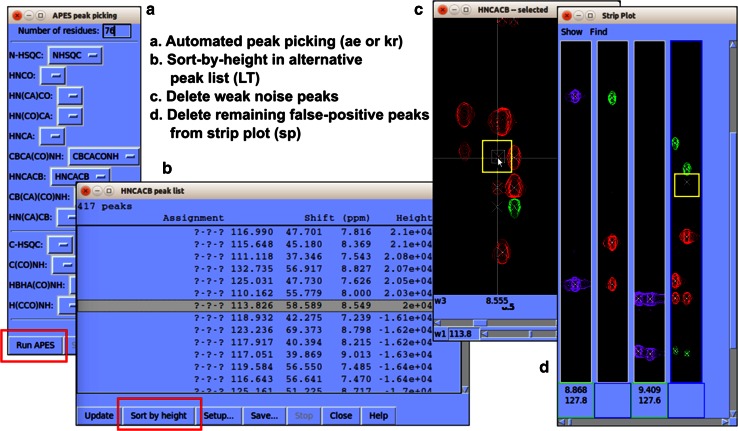
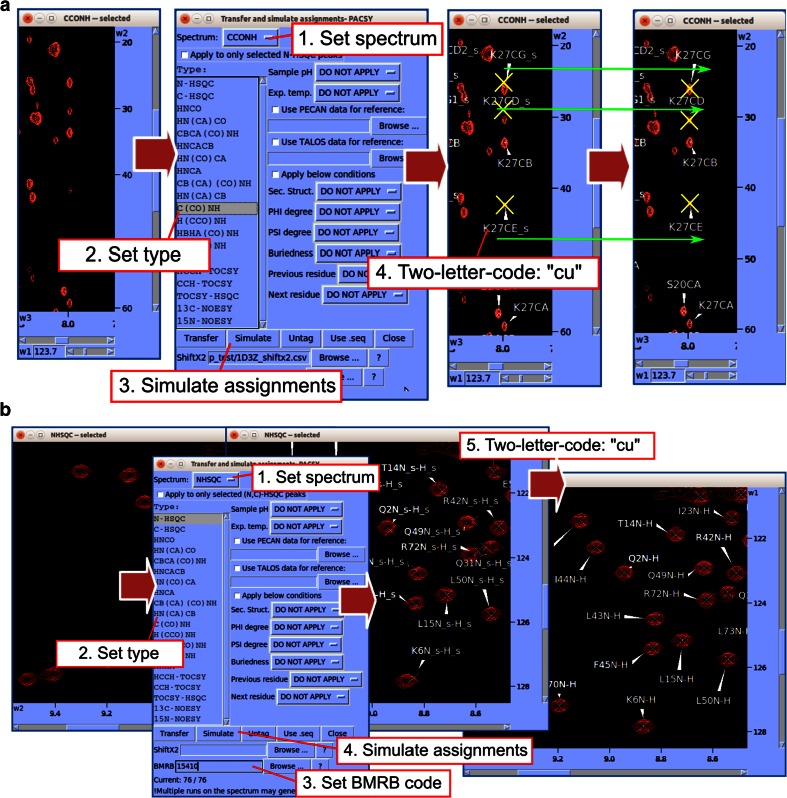
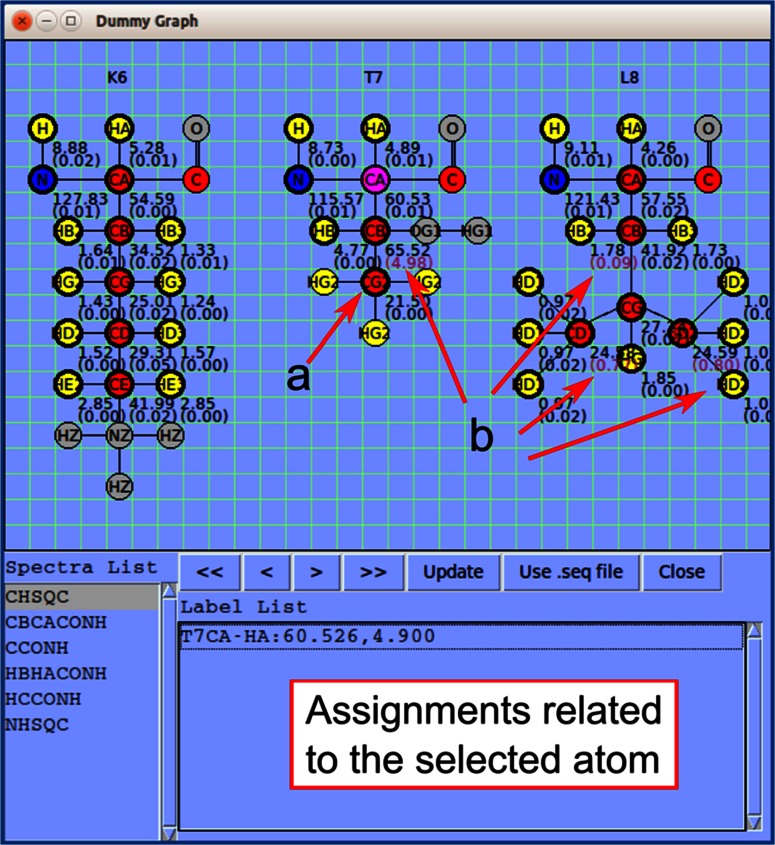
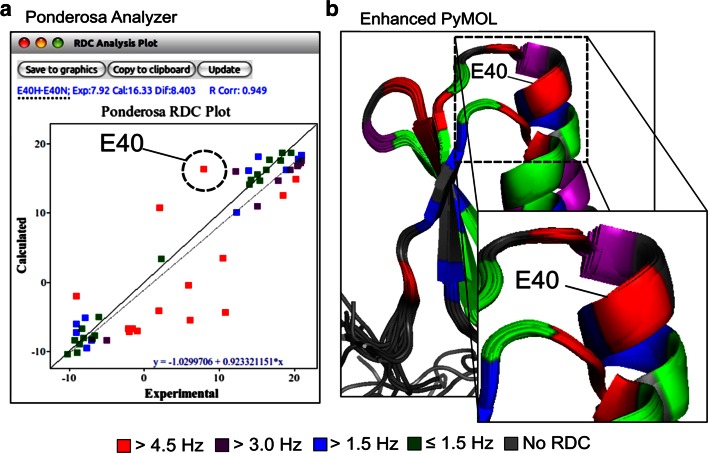
NMR spectroscopy is a powerful technique for determining structural and functional features of biomolecules in physiological solution as well as for observing their intermolecular interactions in real-time. However, complex steps associated with its practice have made the approach daunting for non-specialists. We introduce an NMR platform that makes biomolecular NMR spectroscopy much more accessible by integrating tools, databases, web services, and video tutorials that can be launched by simple installation of NMRFAM software packages or using a cross-platform virtual machine that can be run on any standard laptop or desktop computer. The software package can be downloaded freely from the NMRFAM software download page ( http://pine.nmrfam.wisc.edu/download_packages.html ), and detailed instructions are available from the Integrative NMR Video Tutorial page ( http://pine.nmrfam.wisc.edu/integrative.html ).
Keywords: Automated spectral analysis; Automated structure determination and validation; Chemical shift assignment and validation; Peak identification; Restraint visualization and validation; Visualization of spectra, assignments, and structures.
Figures
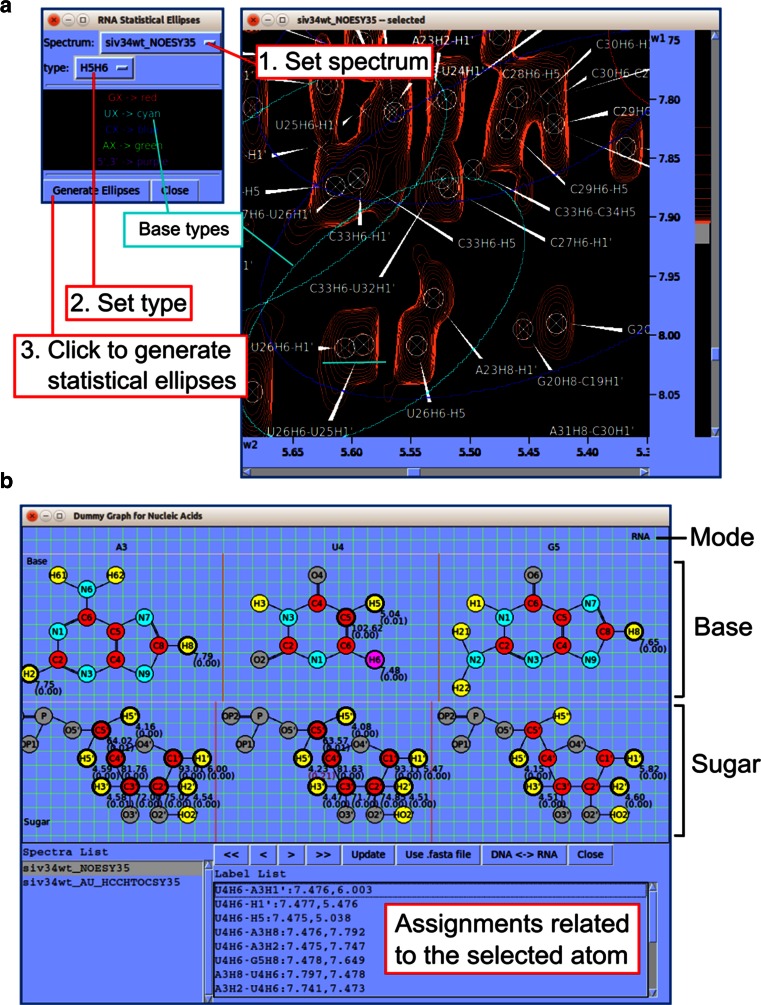
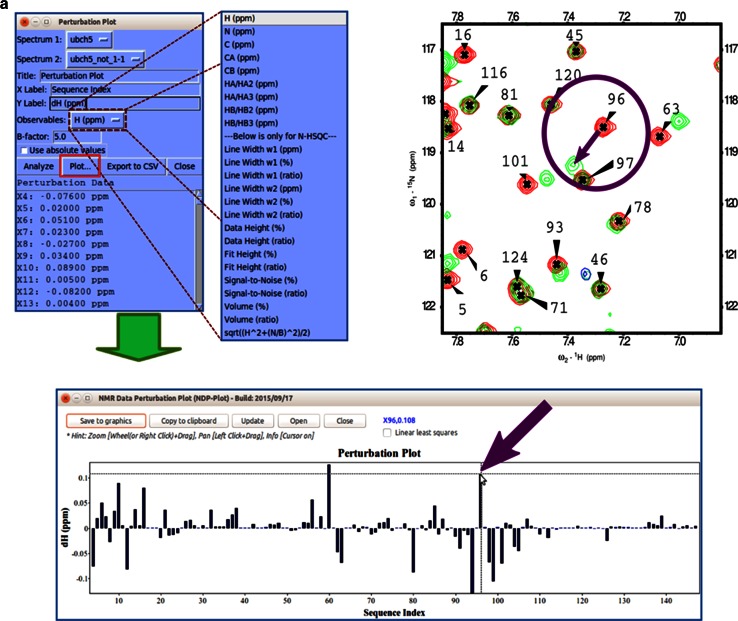
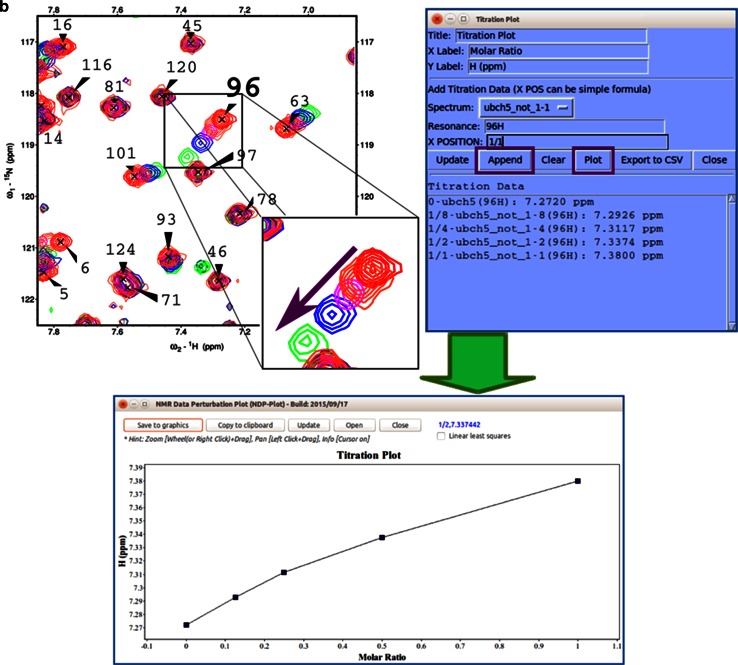
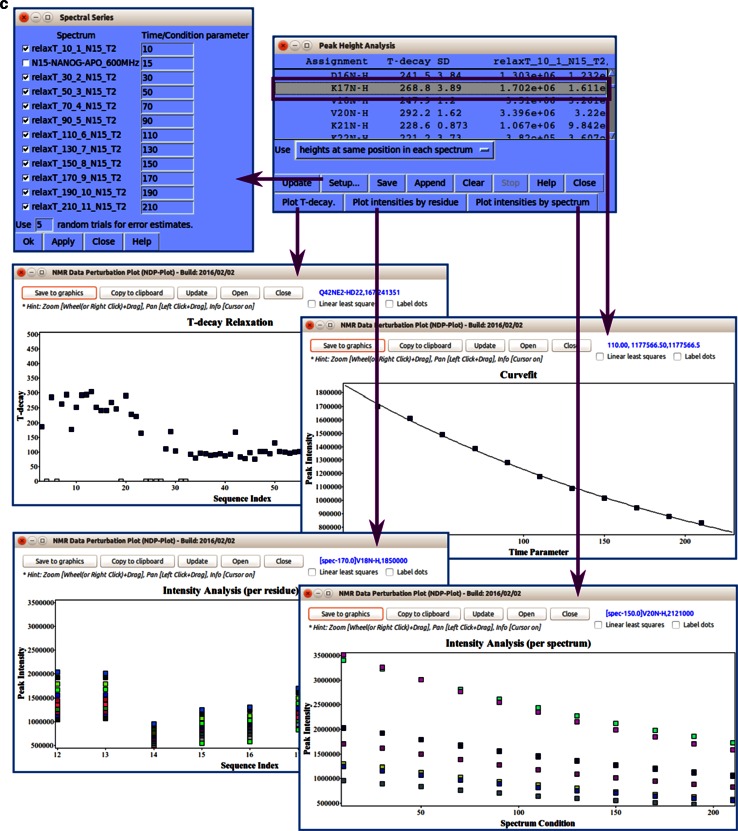
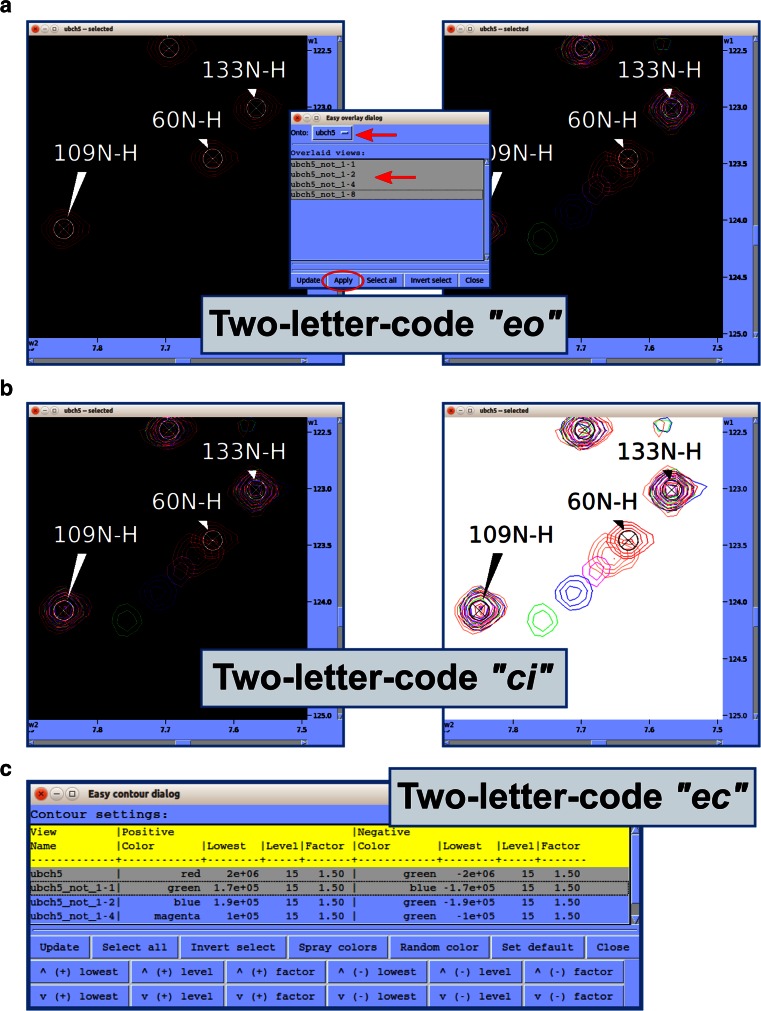
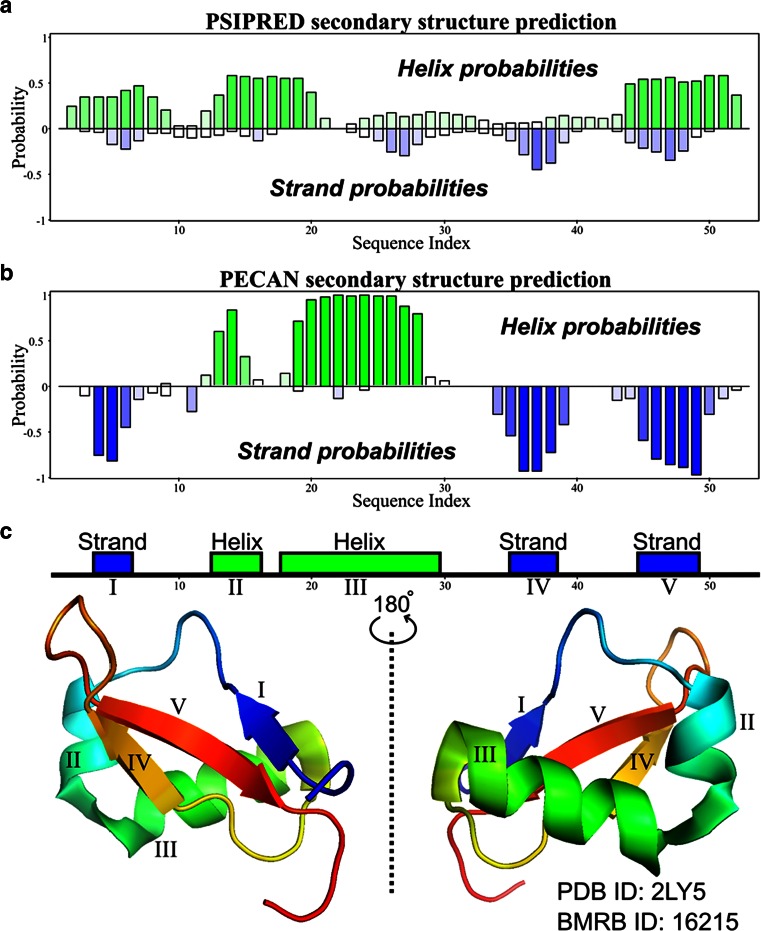
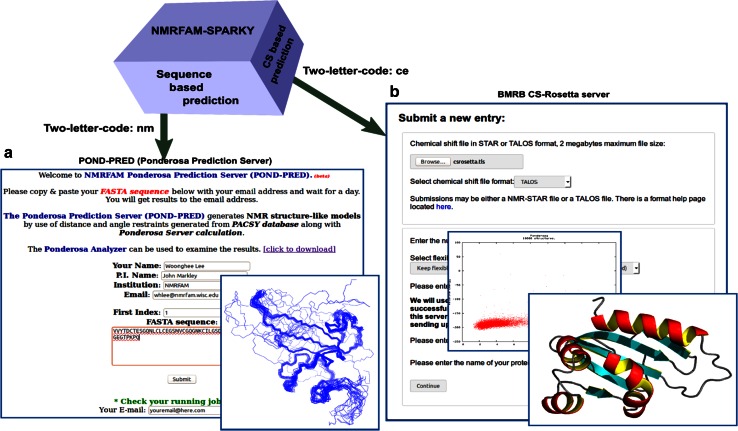
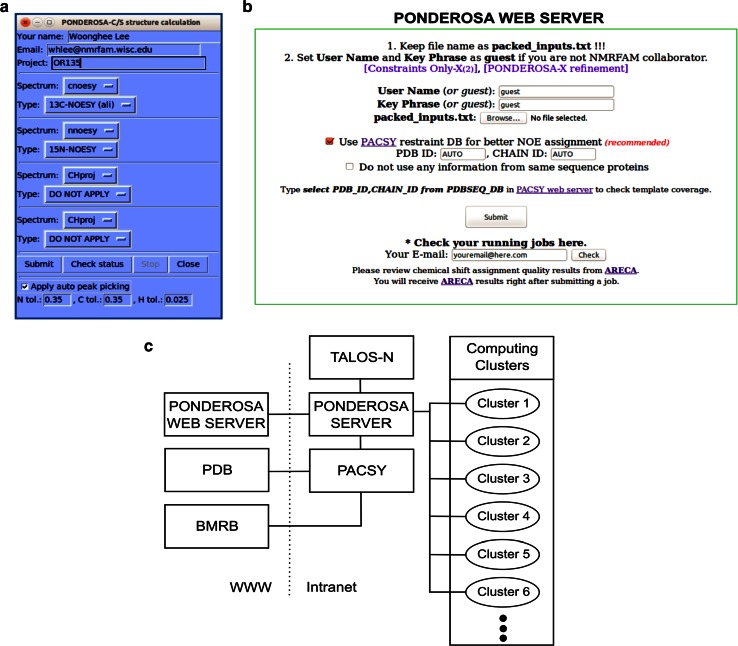
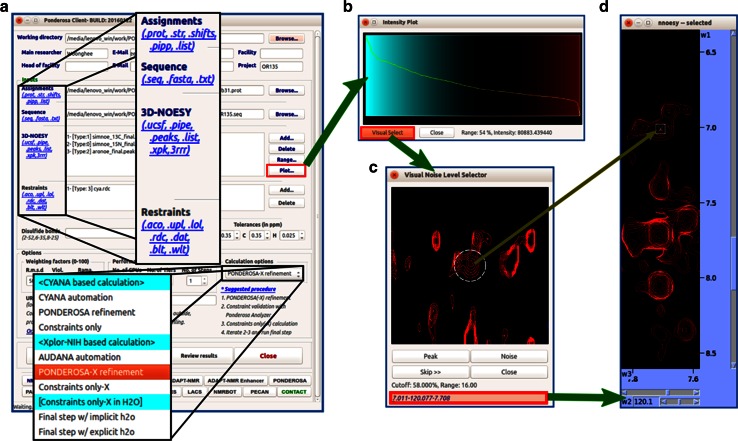
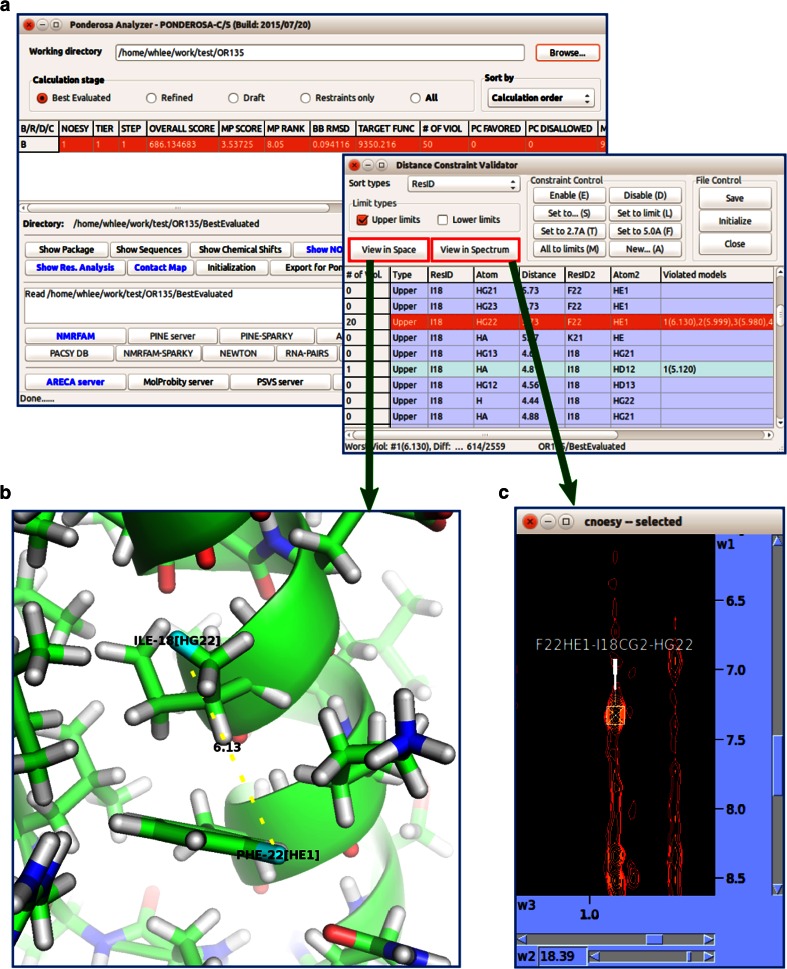
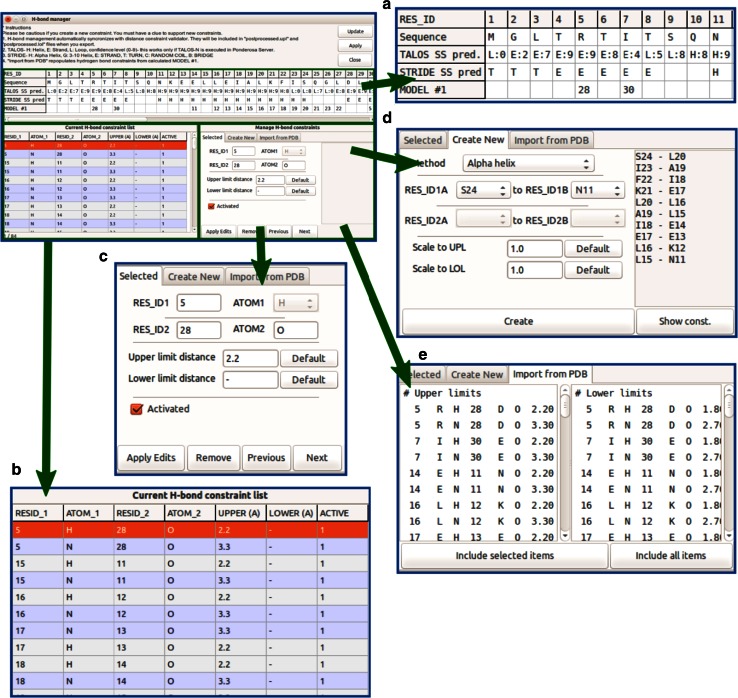
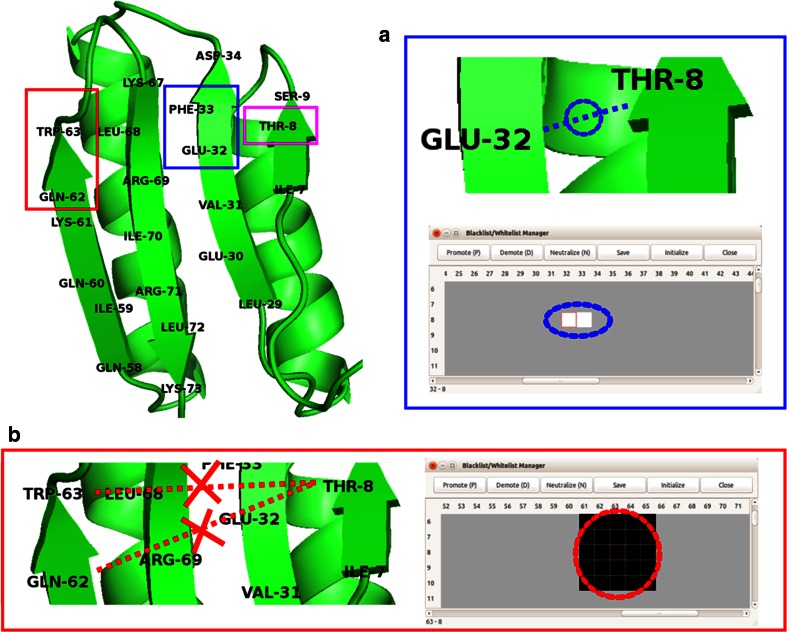
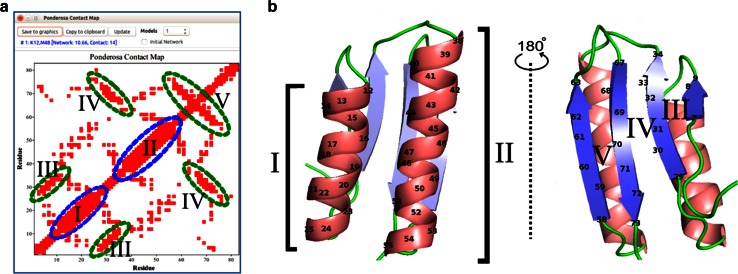

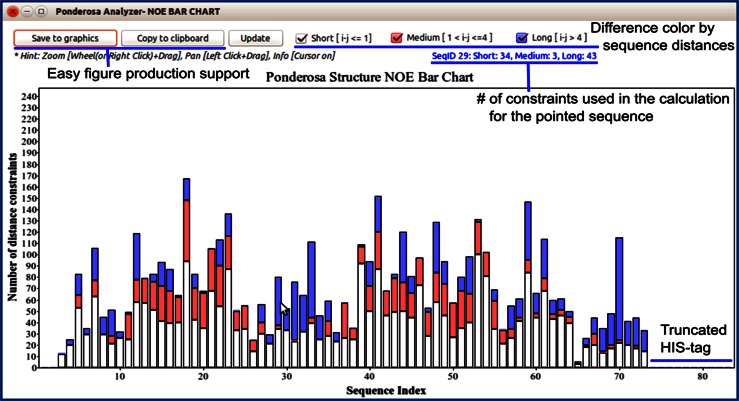
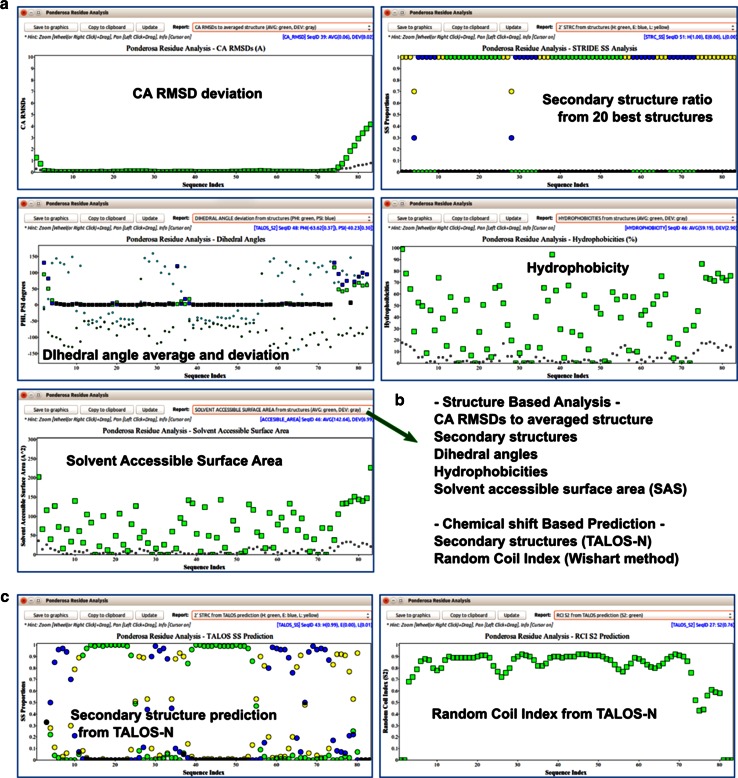
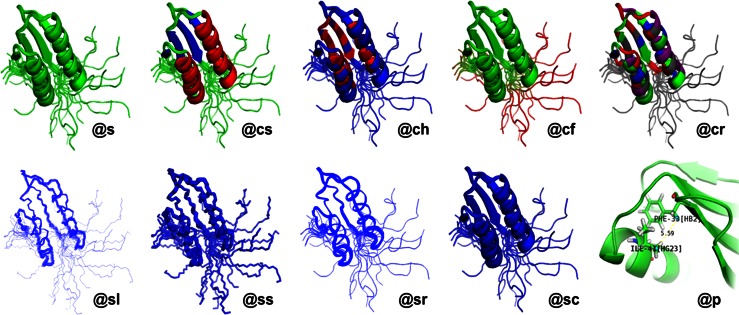
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
