

Emergence of a Large-Plaque Variant in Mice Infected with Coxsackievirus B3
- PMID: 27025249
- PMCID: PMC4817249
- DOI: 10.1128/mBio.00119-16
Emergence of a Large-Plaque Variant in Mice Infected with Coxsackievirus B3
Abstract
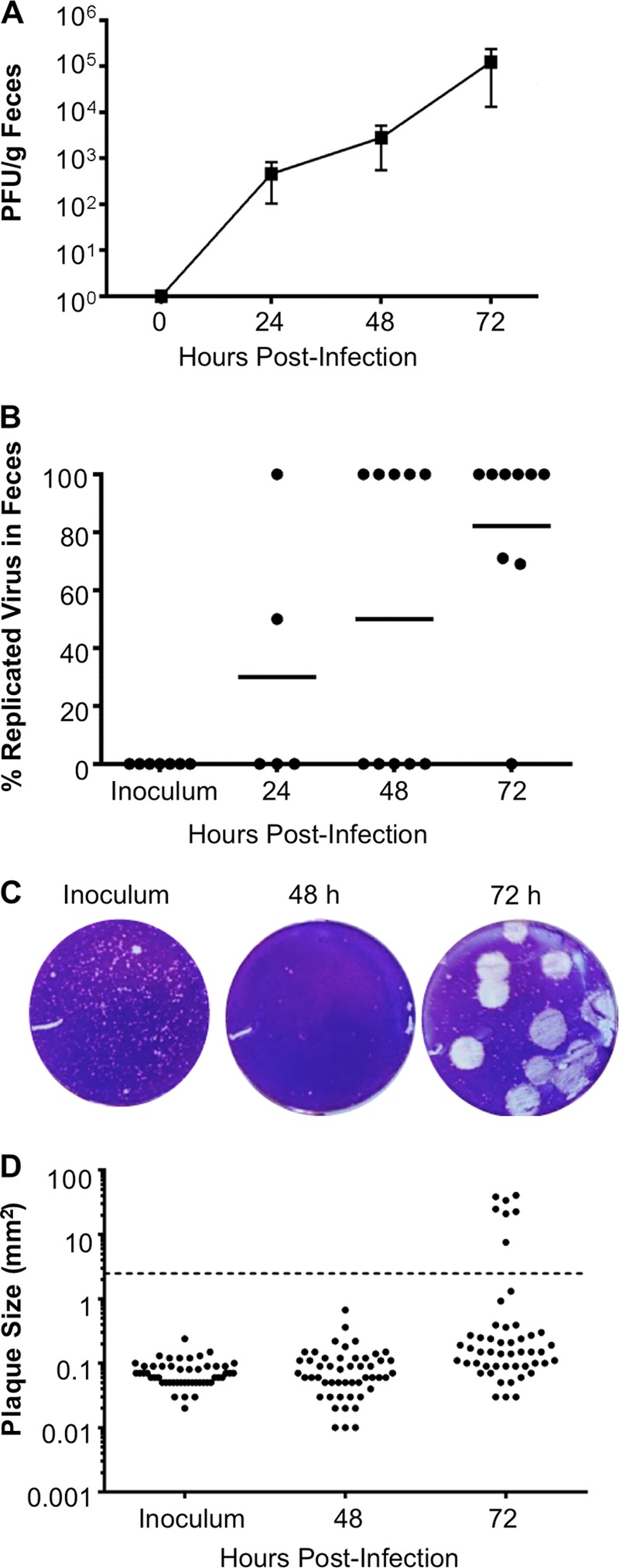
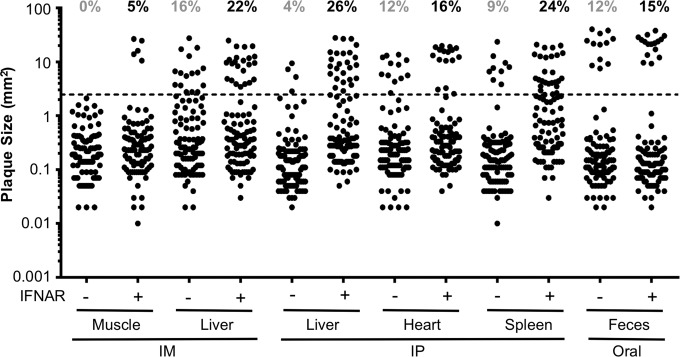
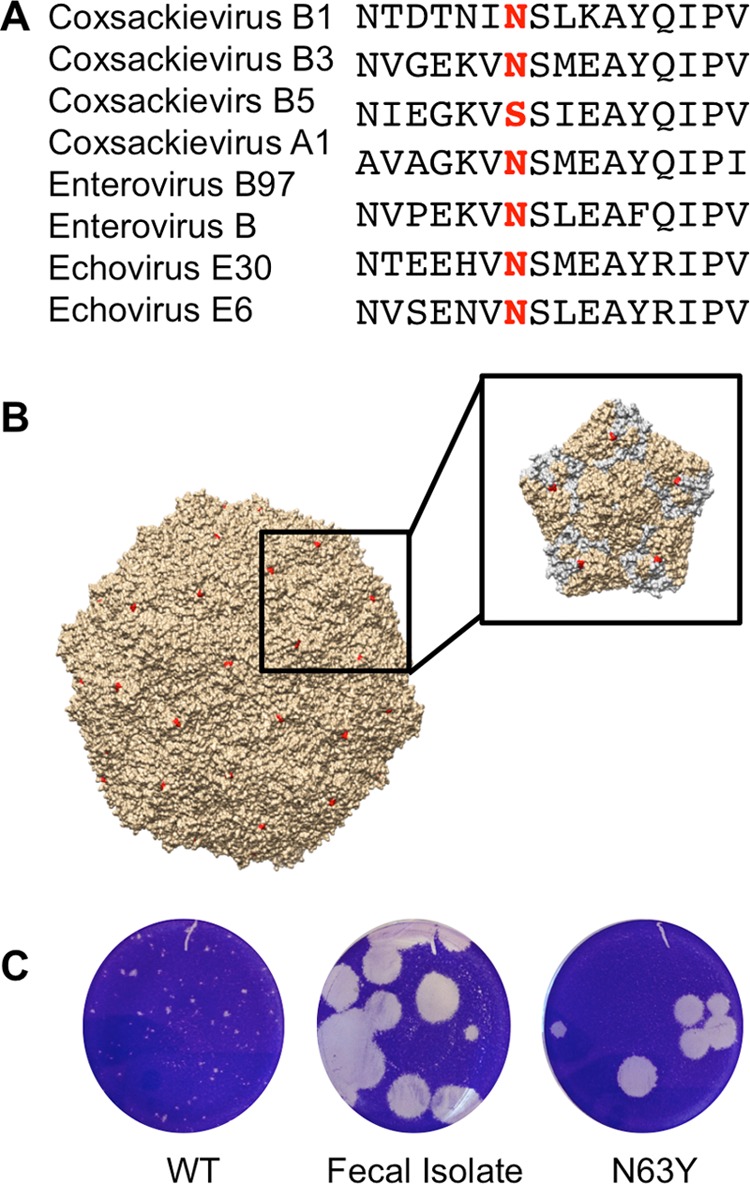
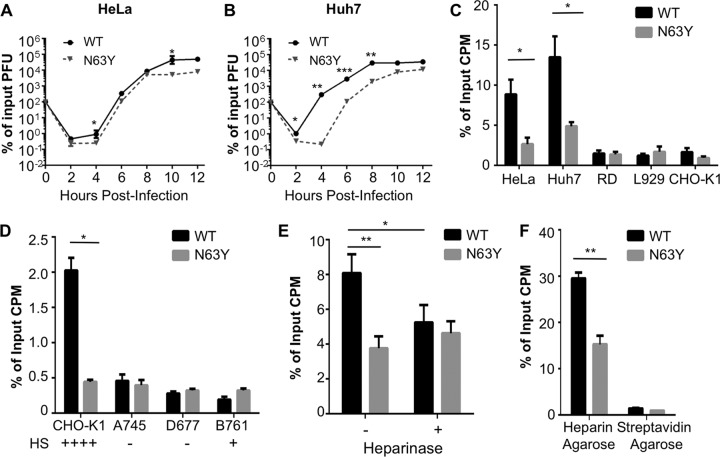
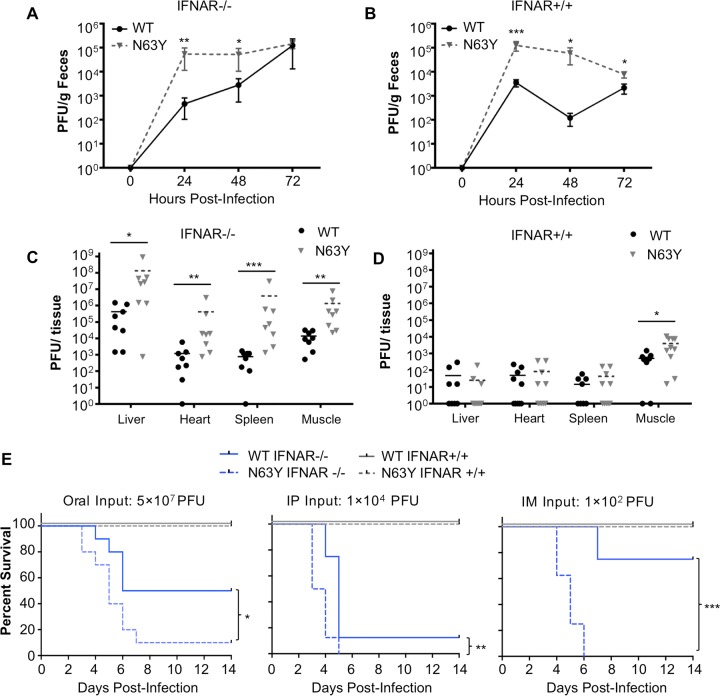
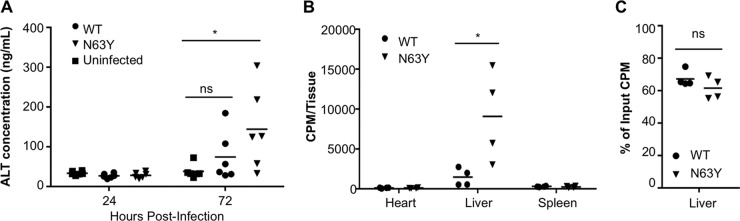
Coxsackieviruses are enteric viruses that frequently infect humans. To examine coxsackievirus pathogenesis, we orally inoculated mice with the coxsackievirus B3 (CVB3) Nancy strain. Using HeLa cell plaque assays with agar overlays, we noticed that some fecal viruses generated plaques >100 times as large as inoculum viruses. These large-plaque variants emerged following viral replication in several different tissues. We identified a single amino acid change, N63Y, in the VP3 capsid protein that was sufficient to confer the large-plaque phenotype. Wild-type CVB3 and N63Y mutant CVB3 had similar plaque sizes when agarose was used in the overlay instead of agar. We determined that sulfated glycans in agar inhibited plaque formation by wild-type CVB3 but not by N63Y mutant CVB3. Furthermore, N63Y mutant CVB3 bound heparin, a sulfated glycan, less efficiently than wild-type CVB3 did. While N63Y mutant CVB3 had a growth defect in cultured cells and reduced attachment, it had enhanced replication and pathogenesis in mice. Infection with N63Y mutant CVB3 induced more severe hepatic damage than infection with wild-type CVB3, likely because N63Y mutant CVB3 disseminates more efficiently to the liver. Our data reinforce the idea that culture-adapted laboratory virus strains can have reduced fitnessin vivo N63Y mutant CVB3 may be useful as a platform to understand viral adaptation and pathogenesis in animal studies.
Importance: Coxsackieviruses frequently infect humans, and although many infections are mild or asymptomatic, there can be severe outcomes, including heart inflammation. Most studies with coxsackieviruses and other viruses use laboratory-adapted viral strains because of their efficient replication in cell culture. We used a cell culture-adapted strain of CVB3, Nancy, to examine viral replication and pathogenesis in orally inoculated mice. We found that mice shed viruses distinct from input viruses because they formed extremely large plaques in cell culture. We identified a single mutation, VP3 N63Y, that was sufficient for large-plaque formation. N63Y mutant viruses have reduced glycan binding and replication in cell culture; however, they have enhanced replication and virulence in mice. We are now using N63Y mutant CVB3 as an improved system for viral pathogenesis studies.
Copyright © 2016 Wang and Pfeiffer.
Figures
Similar articles
-
A mutation in the puff region of VP2 attenuates the myocarditic phenotype of an infectious cDNA of the Woodruff variant of coxsackievirus B3.J Virol. 1996 Nov;70(11):7811-8. doi: 10.1128/JVI.70.11.7811-7818.1996. J Virol. 1996. PMID: 8892902 Free PMC article.
-
Unraveling the molecular basis of membrane-associated release of coxsackievirus B3.Sci Rep. 2025 Mar 10;15(1):8314. doi: 10.1038/s41598-025-92289-x. Sci Rep. 2025. PMID: 40064995 Free PMC article.
-
A single amino acid substitution in the capsid protein VP1 of coxsackievirus B3 (CVB3) alters plaque phenotype in Vero cells but not cardiovirulence in a mouse model.Arch Virol. 1995;140(5):959-66. doi: 10.1007/BF01314972. Arch Virol. 1995. PMID: 7605207
-
A Kidnapping Story: How Coxsackievirus B3 and Its Host Cell Interact.Cell Physiol Biochem. 2019;53(1):121-140. doi: 10.33594/000000125. Cell Physiol Biochem. 2019. PMID: 31230428 Review.
-
Amending Koch's postulates for viral disease: When "growth in pure culture" leads to a loss of virulence.Antiviral Res. 2017 Jan;137:1-5. doi: 10.1016/j.antiviral.2016.11.002. Epub 2016 Nov 8. Antiviral Res. 2017. PMID: 27832942 Free PMC article. Review.
Cited by
-
Bacterial Stabilization of a Panel of Picornaviruses.mSphere. 2019 Apr 3;4(2):e00183-19. doi: 10.1128/mSphere.00183-19. mSphere. 2019. PMID: 30944213 Free PMC article.
-
Coxsackievirus group B3 regulates ASS1-mediated metabolic reprogramming and promotes macrophage inflammatory polarization in viral myocarditis.J Virol. 2024 Sep 17;98(9):e0080524. doi: 10.1128/jvi.00805-24. Epub 2024 Aug 28. J Virol. 2024. PMID: 39194244 Free PMC article.
-
Pathogens that infect mammalian cells via sulfonated glycosaminoglycans.Front Cell Infect Microbiol. 2025 Jun 10;15:1613923. doi: 10.3389/fcimb.2025.1613923. eCollection 2025. Front Cell Infect Microbiol. 2025. PMID: 40557318 Free PMC article. Review.
-
Tradeoffs for a viral mutant with enhanced replication speed.Proc Natl Acad Sci U S A. 2021 Jul 27;118(30):e2105288118. doi: 10.1073/pnas.2105288118. Proc Natl Acad Sci U S A. 2021. PMID: 34282021 Free PMC article.
-
Sex-Dependent Intestinal Replication of an Enteric Virus.J Virol. 2017 Mar 13;91(7):e02101-16. doi: 10.1128/JVI.02101-16. Print 2017 Apr 1. J Virol. 2017. PMID: 28100612 Free PMC article.
References
-
- Tracy S, Drescher KM, Chapman NM, Kim KS, Carson SD, Pirruccello S, Lane PH, Romero JR, Leser JS. 2002. Toward testing the hypothesis that group B coxsackieviruses (CVB) trigger insulin-dependent diabetes: inoculating nonobese diabetic mice with CVB markedly lowers diabetes incidence. J Virol 76:12097–12111. doi:10.1128/JVI.76.23.12097-12111.2002. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
