

Murine startle mutant Nmf11 affects the structural stability of the glycine receptor and increases deactivation
- PMID: 27028707
- PMCID: PMC4929309
- DOI: 10.1113/JP272122
Murine startle mutant Nmf11 affects the structural stability of the glycine receptor and increases deactivation
Abstract
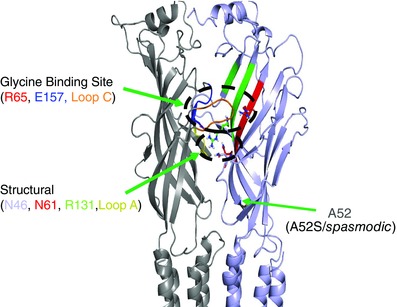
Key points: Hyperekplexia or startle disease is a serious neurological condition affecting newborn children and usually involves dysfunctional glycinergic neurotransmission. Glycine receptors (GlyRs) are major mediators of inhibition in the spinal cord and brainstem. A missense mutation, replacing asparagine (N) with lysine (K), at position 46 in the GlyR α1 subunit induced hyperekplexia following a reduction in the potency of the transmitter glycine; this resulted from a rapid deactivation of the agonist current at mutant GlyRs. These effects of N46K were rescued by mutating a juxtaposed residue, N61 on binding Loop D, suggesting these two asparagines may interact. Asparagine 46 is considered to be important for the structural stability of the subunit interface and glycine binding site, and its mutation represents a new mechanism by which GlyR dysfunction induces startle disease.
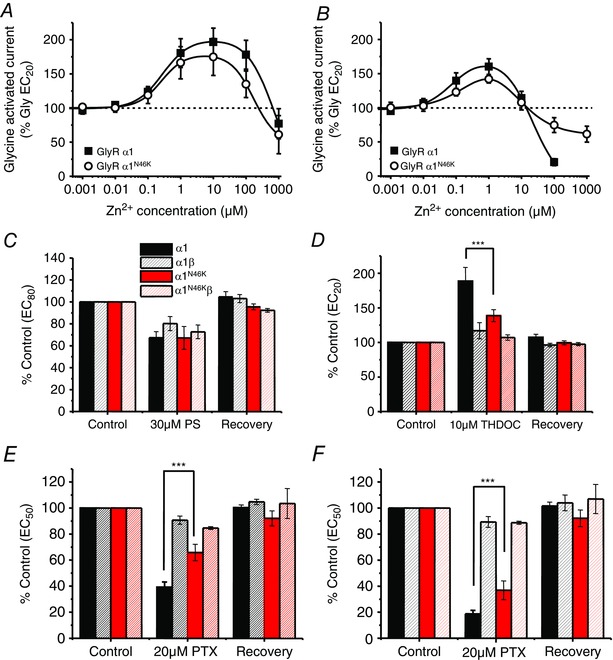
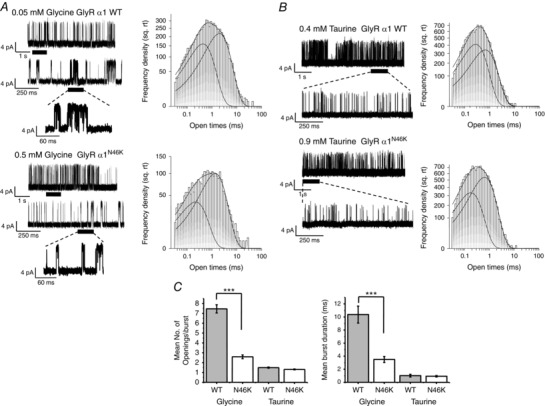
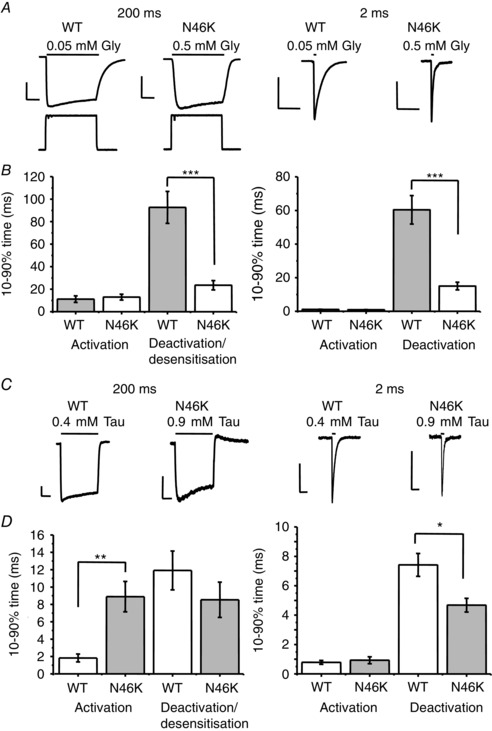
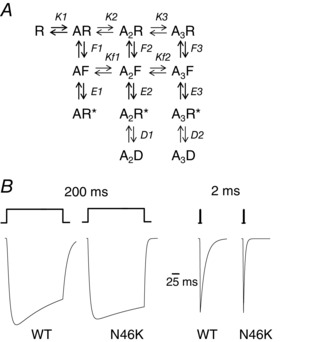
Abstract: Dysfunctional glycinergic inhibitory transmission underlies the debilitating neurological condition, hyperekplexia, which is characterised by exaggerated startle reflexes, muscle hypertonia and apnoea. Here we investigated the N46K missense mutation in the GlyR α1 subunit gene found in the ethylnitrosourea (ENU) murine mutant, Nmf11, which causes reduced body size, evoked tremor, seizures, muscle stiffness, and morbidity by postnatal day 21. Introducing the N46K mutation into recombinant GlyR α1 homomeric receptors, expressed in HEK cells, reduced the potencies of glycine, β-alanine and taurine by 9-, 6- and 3-fold respectively, and that of the competitive antagonist strychnine by 15-fold. Replacing N46 with hydrophobic, charged or polar residues revealed that the amide moiety of asparagine was crucial for GlyR activation. Co-mutating N61, located on a neighbouring β loop to N46, rescued the wild-type phenotype depending on the amino acid charge. Single-channel recording identified that burst length for the N46K mutant was reduced and fast agonist application revealed faster glycine deactivation times for the N46K mutant compared with the WT receptor. Overall, these data are consistent with N46 ensuring correct alignment of the α1 subunit interface by interaction with juxtaposed residues to preserve the structural integrity of the glycine binding site. This represents a new mechanism by which GlyR dysfunction induces startle disease.
© 2016 The Authors. The Journal of Physiology published by John Wiley & Sons Ltd on behalf of The Physiological Society.
Figures
References
-
- Belelli D, Pistis M, Peters JA & Lambert JJ (1999). The interaction of general anaesthetics and neurosteroids with GABAA and glycine receptors. Neurochem Int 34, 447–452. - PubMed
-
- Bloomenthal AB, Goldwater E, Pritchett DB & Harrison NL (1994). Biphasic modulation of the strychnine‐sensitive glycine receptor by Zn2+ . Mol Pharmacol 46, 1156–1159. - PubMed
-
- Brejc K, van Dijk WJ, Smit AB & Sixma TK (2002). The 2.7 A structure of AChBP, homologue of the ligand‐binding domain of the nicotinic acetylcholine receptor. Novartis Found Symp 245, 22–29. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous