

Somatic PIK3CA mutations as a driver of sporadic venous malformations
- PMID: 27030594
- PMCID: PMC4962922
- DOI: 10.1126/scitranslmed.aaf1164
Somatic PIK3CA mutations as a driver of sporadic venous malformations
Abstract
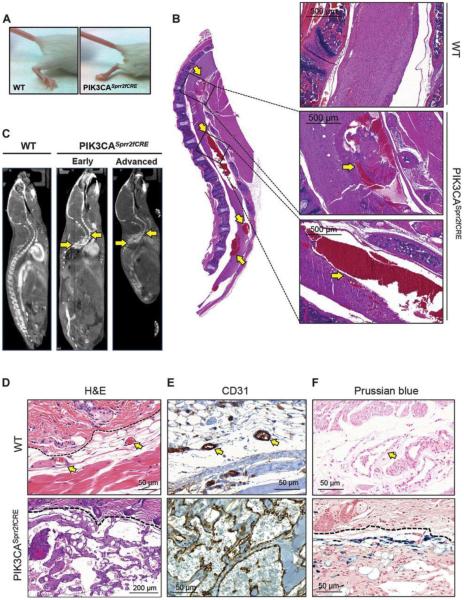
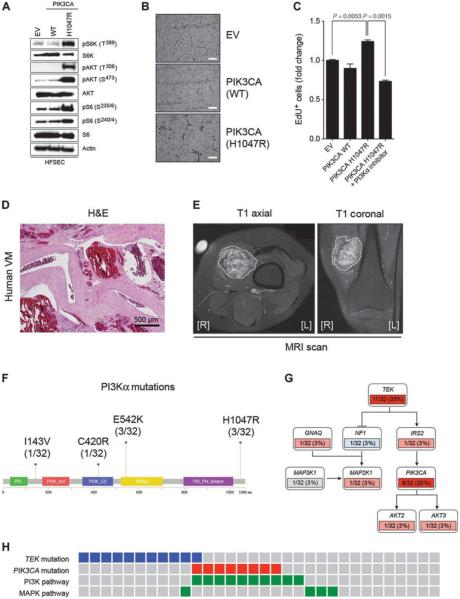
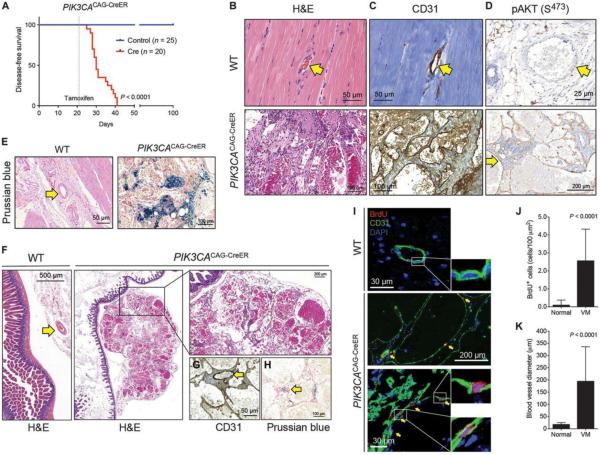
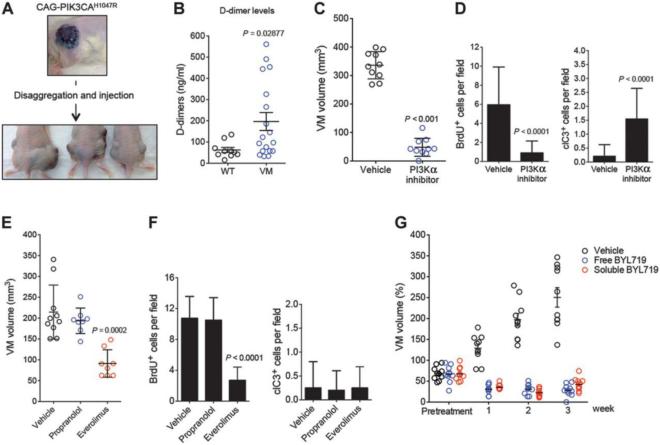
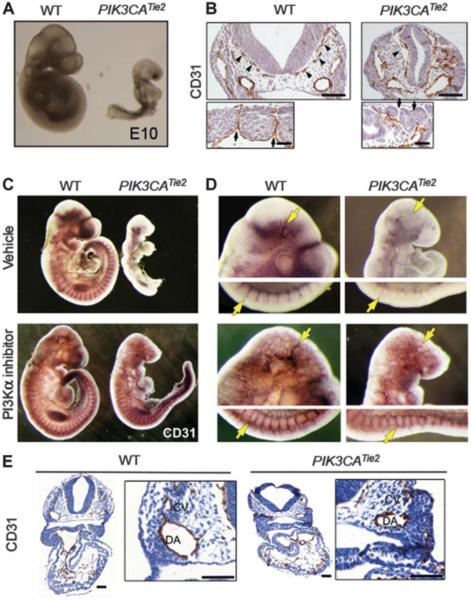
Venous malformations (VM) are vascular malformations characterized by enlarged and distorted blood vessel channels. VM grow over time and cause substantial morbidity because of disfigurement, bleeding, and pain, representing a clinical challenge in the absence of effective treatments (Nguyenet al, 2014; Uebelhoeret al, 2012). Somatic mutations may act as drivers of these lesions, as suggested by the identification of TEK mutations in a proportion of VM (Limayeet al, 2009). We report that activating PIK3CA mutations gives rise to sporadic VM in mice, which closely resemble the histology of the human disease. Furthermore, we identified mutations in PIK3CA and related genes of the PI3K (phosphatidylinositol 3-kinase)/AKT pathway in about 30% of human VM that lack TEK alterations. PIK3CA mutations promote downstream signaling and proliferation in endothelial cells and impair normal vasculogenesis in embryonic development. We successfully treated VM in mouse models using pharmacological inhibitors of PI3Kα administered either systemically or topically. This study elucidates the etiology of a proportion of VM and proposes a therapeutic approach for this disease.
Copyright © 2016, American Association for the Advancement of Science.
Figures
Similar articles
-
Somatic activating mutations in Pik3ca cause sporadic venous malformations in mice and humans.Sci Transl Med. 2016 Mar 30;8(332):332ra43. doi: 10.1126/scitranslmed.aad9982. Sci Transl Med. 2016. PMID: 27030595 Free PMC article.
-
PI3K/mTOR inhibition promotes the regression of experimental vascular malformations driven by PIK3CA-activating mutations.Cell Death Dis. 2018 Jan 19;9(2):45. doi: 10.1038/s41419-017-0064-x. Cell Death Dis. 2018. PMID: 29352118 Free PMC article.
-
Somatic Activating PIK3CA Mutations Cause Venous Malformation.Am J Hum Genet. 2015 Dec 3;97(6):914-21. doi: 10.1016/j.ajhg.2015.11.011. Am J Hum Genet. 2015. PMID: 26637981 Free PMC article.
-
Genetic landscape of common venous malformations in the head and neck.J Vasc Surg Venous Lymphat Disord. 2021 Jul;9(4):1007-1016.e7. doi: 10.1016/j.jvsv.2020.11.016. Epub 2020 Nov 26. J Vasc Surg Venous Lymphat Disord. 2021. PMID: 33248299 Review.
-
PIK3CA mutations in vascular malformations.Curr Opin Hematol. 2019 May;26(3):170-178. doi: 10.1097/MOH.0000000000000496. Curr Opin Hematol. 2019. PMID: 30855339 Review.
Cited by
-
Cutaneous and hepatic vascular lesions due to a recurrent somatic GJA4 mutation reveal a pathway for vascular malformation.HGG Adv. 2021 Apr 8;2(2):100028. doi: 10.1016/j.xhgg.2021.100028. Epub 2021 Mar 1. HGG Adv. 2021. PMID: 33912852 Free PMC article.
-
Light-activated gold nanorods for effective therapy of venous malformation.Mater Today Bio. 2022 Aug 18;16:100401. doi: 10.1016/j.mtbio.2022.100401. eCollection 2022 Dec. Mater Today Bio. 2022. PMID: 36052154 Free PMC article.
-
A Xenograft Model for Venous Malformation.Methods Mol Biol. 2021;2206:179-192. doi: 10.1007/978-1-0716-0916-3_13. Methods Mol Biol. 2021. PMID: 32754818 Free PMC article.
-
Pathophysiology of Slow-Flow Vascular Malformations: Current Understanding and Unanswered Questions.J Vasc Anom (Phila). 2023 Jul 10;4(3):e069. doi: 10.1097/JOVA.0000000000000069. eCollection 2023 Sep. J Vasc Anom (Phila). 2023. PMID: 37662560 Free PMC article. Review.
-
Blockade of VEGF-C signaling inhibits lymphatic malformations driven by oncogenic PIK3CA mutation.Nat Commun. 2020 Jun 8;11(1):2869. doi: 10.1038/s41467-020-16496-y. Nat Commun. 2020. PMID: 32513927 Free PMC article.
References
-
- Brouillard P, Vikkula M. Genetic causes of vascular malformations. Hum. Mol. Gen. 2007;16:R140–R149. - PubMed
-
- Brouillard P, Vikkula M. Vascular malformations: Localized defects in vascular morphogenesis. Clin. Genet. 2003;63:340–351. - PubMed
-
- Nguyen H-L, Boon LM, Vikkula M. Genetics of vascular malformations. Semin. Pediatr. Surg. 2014;23:221–226. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
