

Neuronal heparan sulfates promote amyloid pathology by modulating brain amyloid-β clearance and aggregation in Alzheimer's disease
- PMID: 27030596
- PMCID: PMC5512541
- DOI: 10.1126/scitranslmed.aad3650
Neuronal heparan sulfates promote amyloid pathology by modulating brain amyloid-β clearance and aggregation in Alzheimer's disease
Abstract
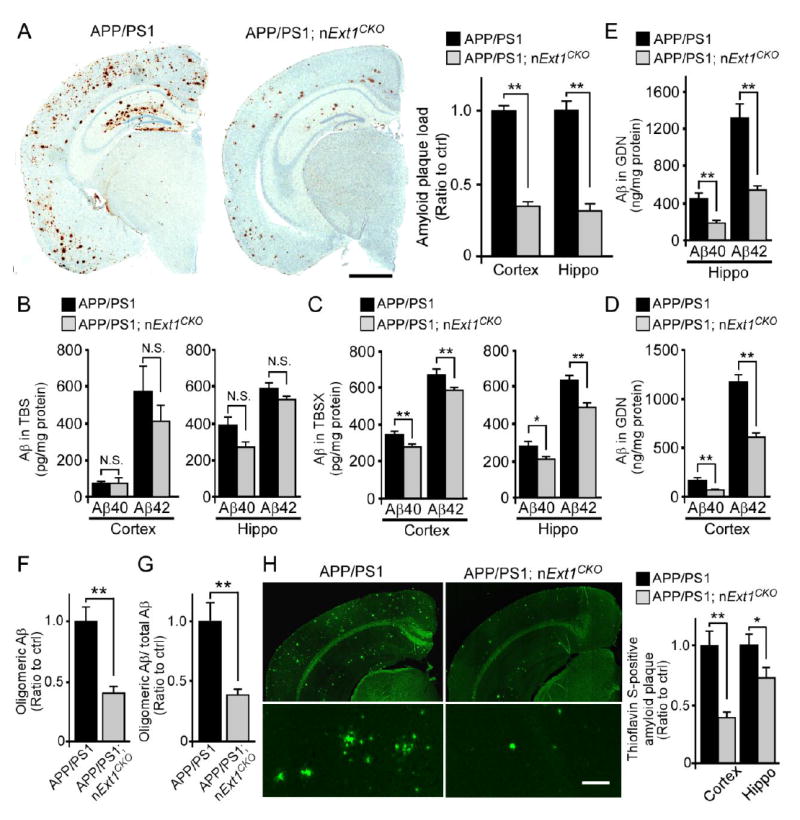
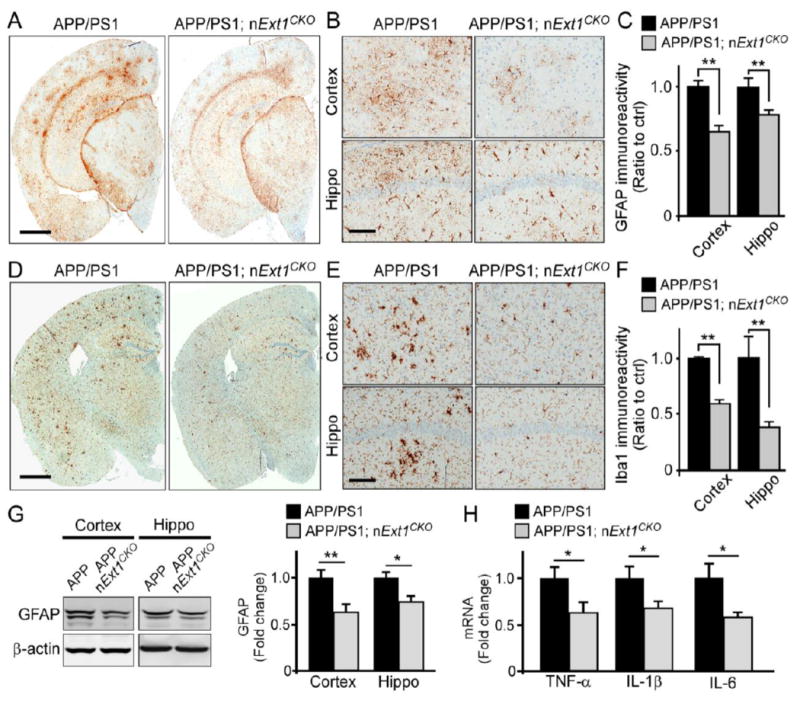
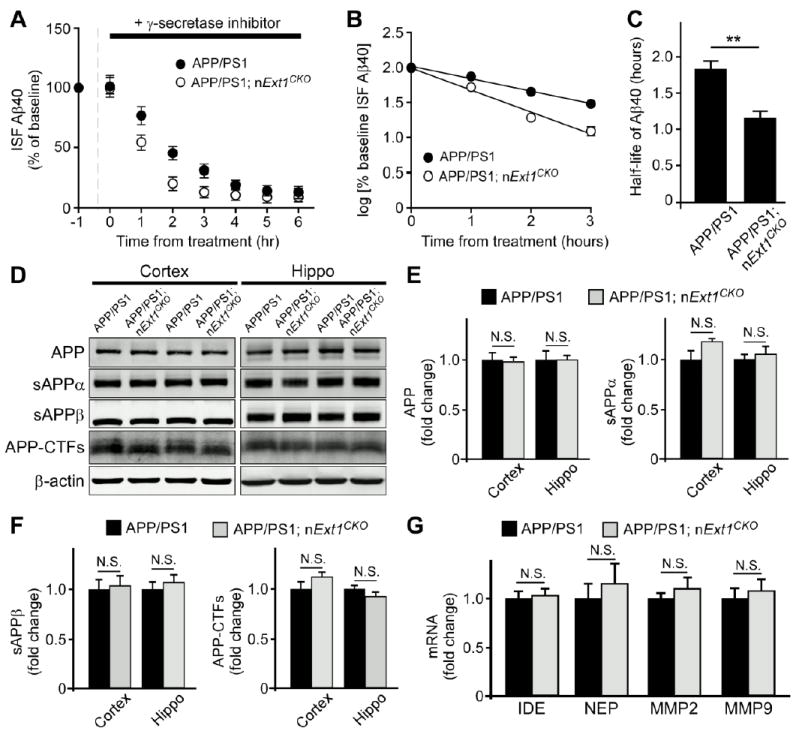
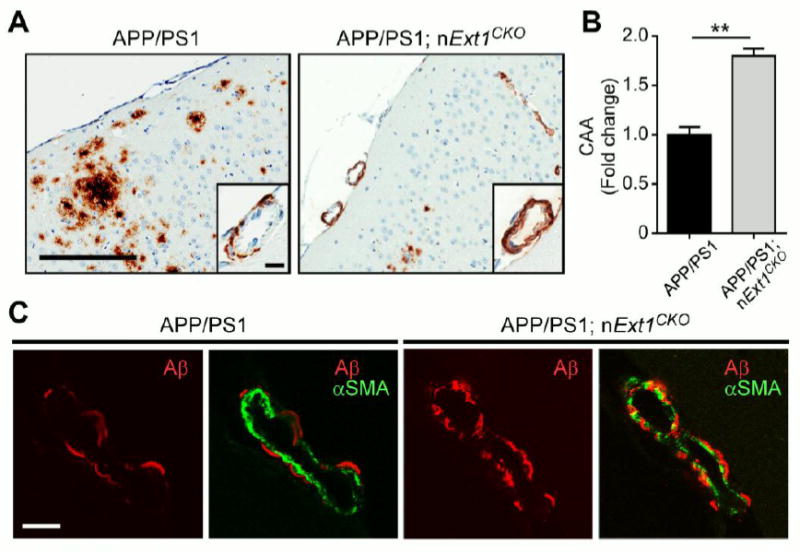
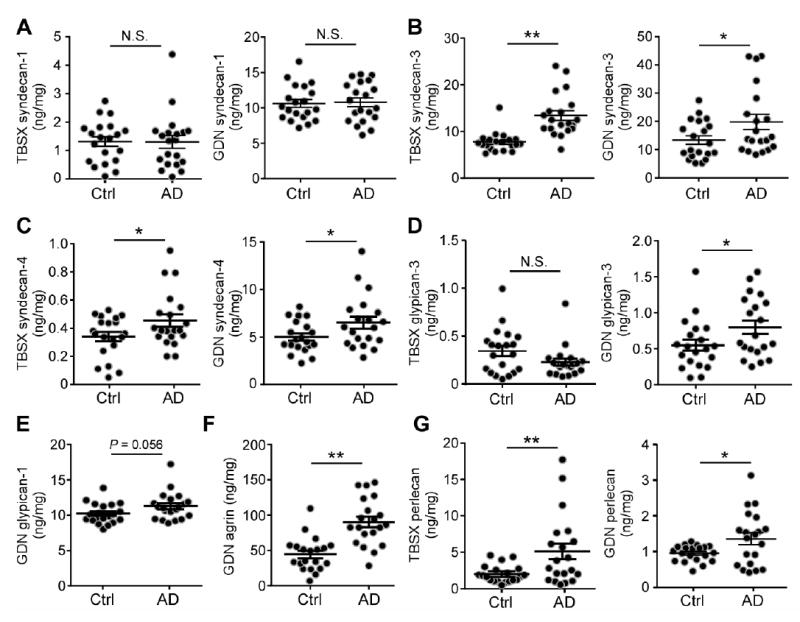
Accumulation of amyloid-β (Aβ) peptide in the brain is the first critical step in the pathogenesis of Alzheimer's disease (AD). Studies in humans suggest that Aβ clearance from the brain is frequently impaired in late-onset AD. Aβ accumulation leads to the formation of Aβ aggregates, which injure synapses and contribute to eventual neurodegeneration. Cell surface heparan sulfates (HSs), expressed on all cell types including neurons, have been implicated in several features in the pathogenesis of AD including its colocalization with amyloid plaques and modulatory role in Aβ aggregation. We show that removal of neuronal HS by conditional deletion of the Ext1 gene, which encodes an essential glycosyltransferase for HS biosynthesis, in postnatal neurons of amyloid model APP/PS1 mice led to a reduction in both Aβ oligomerization and the deposition of amyloid plaques. In vivo microdialysis experiments also detected an accelerated rate of Aβ clearance in the brain interstitial fluid, suggesting that neuronal HS either inhibited or represented an inefficient pathway for Aβ clearance. We found that the amounts of various HS proteoglycans (HSPGs) were increased in postmortem human brain tissues from AD patients, suggesting that this pathway may contribute directly to amyloid pathogenesis. Our findings have implications for AD pathogenesis and provide insight into therapeutic interventions targeting Aβ-HSPG interactions.
Copyright © 2016, American Association for the Advancement of Science.
Conflict of interest statement
Figures
Comment in
-
Heparan Sulfates Promote Amyloid Pathology in Alzheimer Disease.Neurosurgery. 2016 Aug;79(2):N12-3. doi: 10.1227/NEU.0000000000001291. Neurosurgery. 2016. PMID: 27428635 No abstract available.
References
-
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. - PubMed
-
- Vassar R, Bennett BD, Babu-Khan S, Kahn S, Mendiaz EA, Denis P, Teplow DB, Ross S, Amarante P, Loeloff R, Luo Y, Fisher S, Fuller J, Edenson S, Lile J, Jarosinski MA, Biere AL, Curran E, Burgess T, Louis JC, Collins F, Treanor J, Rogers G, Citron M. Beta-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science. 1999;286:735–741. - PubMed
-
- Goate A. Segregation of a missense mutation in the amyloid beta-protein precursor gene with familial Alzheimer’s disease. J Alzheimers Dis. 2006;9:341–347. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- P30 AG028383/AG/NIA NIH HHS/United States
- P50AG016574/AG/NIA NIH HHS/United States
- R01AG046205/AG/NIA NIH HHS/United States
- P01 NS074969/NS/NINDS NIH HHS/United States
- R01 AG027924/AG/NIA NIH HHS/United States
- R01AG027924/AG/NIA NIH HHS/United States
- R01 AG046205/AG/NIA NIH HHS/United States
- R01AG035355/AG/NIA NIH HHS/United States
- P50 AG016574/AG/NIA NIH HHS/United States
- R01 NS088496/NS/NINDS NIH HHS/United States
- R01 AG042513/AG/NIA NIH HHS/United States
- RF1 AG051504/AG/NIA NIH HHS/United States
- R01 AG035355/AG/NIA NIH HHS/United States
- R01NS088496/NS/NINDS NIH HHS/United States
- P01NS074969/NS/NINDS NIH HHS/United States
- R01AG042513/AG/NIA NIH HHS/United States
- RF1 AG046205/AG/NIA NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
