

Structural and Genetic Studies Demonstrate Neurologic Dysfunction in Triosephosphate Isomerase Deficiency Is Associated with Impaired Synaptic Vesicle Dynamics
- PMID: 27031109
- PMCID: PMC4816394
- DOI: 10.1371/journal.pgen.1005941
Structural and Genetic Studies Demonstrate Neurologic Dysfunction in Triosephosphate Isomerase Deficiency Is Associated with Impaired Synaptic Vesicle Dynamics
Abstract
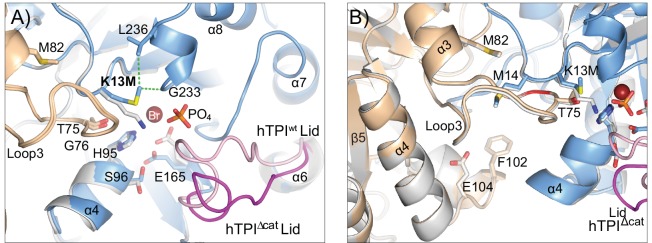
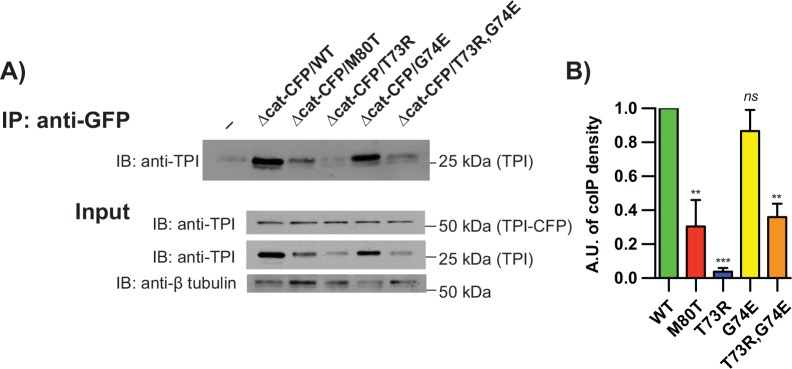
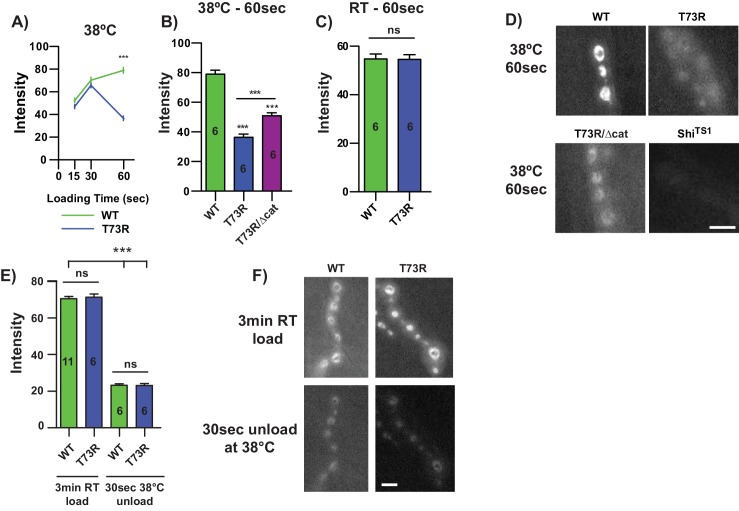
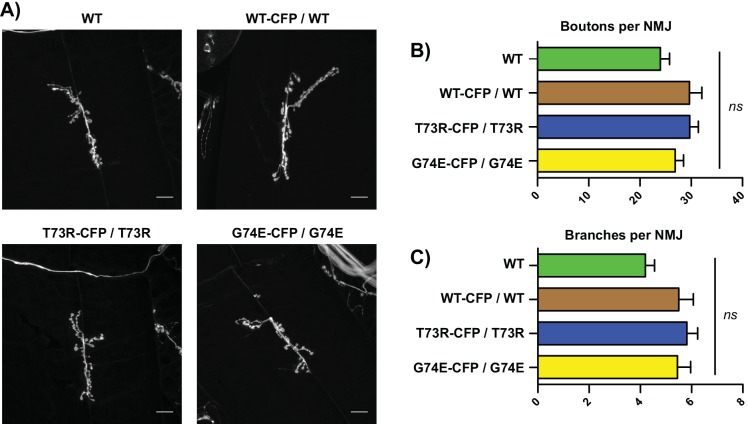
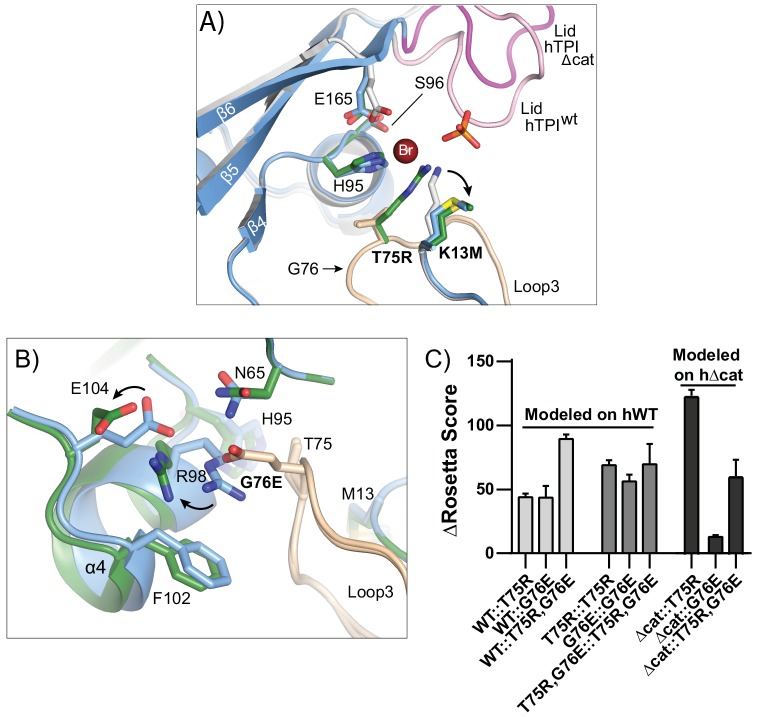
Triosephosphate isomerase (TPI) deficiency is a poorly understood disease characterized by hemolytic anemia, cardiomyopathy, neurologic dysfunction, and early death. TPI deficiency is one of a group of diseases known as glycolytic enzymopathies, but is unique for its severe patient neuropathology and early mortality. The disease is caused by missense mutations and dysfunction in the glycolytic enzyme, TPI. Previous studies have detailed structural and catalytic changes elicited by disease-associated TPI substitutions, and samples of patient erythrocytes have yielded insight into patient hemolytic anemia; however, the neuropathophysiology of this disease remains a mystery. This study combines structural, biochemical, and genetic approaches to demonstrate that perturbations of the TPI dimer interface are sufficient to elicit TPI deficiency neuropathogenesis. The present study demonstrates that neurologic dysfunction resulting from TPI deficiency is characterized by synaptic vesicle dysfunction, and can be attenuated with catalytically inactive TPI. Collectively, our findings are the first to identify, to our knowledge, a functional synaptic defect in TPI deficiency derived from molecular changes in the TPI dimer interface.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
Similar articles
-
Missense variant in TPI1 (Arg189Gln) causes neurologic deficits through structural changes in the triosephosphate isomerase catalytic site and reduced enzyme levels in vivo.Biochim Biophys Acta Mol Basis Dis. 2019 Sep 1;1865(9):2257-2266. doi: 10.1016/j.bbadis.2019.05.002. Epub 2019 May 7. Biochim Biophys Acta Mol Basis Dis. 2019. PMID: 31075491 Free PMC article.
-
Triosephosphate isomerase I170V alters catalytic site, enhances stability and induces pathology in a Drosophila model of TPI deficiency.Biochim Biophys Acta. 2015 Jan;1852(1):61-9. doi: 10.1016/j.bbadis.2014.10.010. Epub 2014 Oct 16. Biochim Biophys Acta. 2015. PMID: 25463631 Free PMC article.
-
Triosephosphate isomerase deficiency: Effect of F240L mutation on enzyme structure.Arch Biochem Biophys. 2020 Aug 15;689:108473. doi: 10.1016/j.abb.2020.108473. Epub 2020 Jun 22. Arch Biochem Biophys. 2020. PMID: 32585311
-
Triosephosphate isomerase deficiency: facts and doubts.IUBMB Life. 2006 Dec;58(12):703-15. doi: 10.1080/15216540601115960. IUBMB Life. 2006. PMID: 17424909 Review.
-
The feasibility of replacement therapy for inherited disorder of glycolysis: triosephosphate isomerase deficiency (review).Int J Mol Med. 1998 Dec;2(6):701-4. doi: 10.3892/ijmm.2.6.701. Int J Mol Med. 1998. PMID: 9850739 Review.
Cited by
-
Newly discovered roles of triosephosphate isomerase including functions within the nucleus.Mol Med. 2023 Jan 31;29(1):18. doi: 10.1186/s10020-023-00612-x. Mol Med. 2023. PMID: 36721084 Free PMC article. Review.
-
Aging, Alzheimer's Disease and Dysfunctional Glycolysis; Similar Effects of Too Much and Too Little.Aging Dis. 2019 Dec 1;10(6):1328-1331. doi: 10.14336/AD.2019.0611. eCollection 2019 Dec. Aging Dis. 2019. PMID: 31788344 Free PMC article.
-
Presynaptic disorders: a clinical and pathophysiological approach focused on the synaptic vesicle.J Inherit Metab Dis. 2018 Nov;41(6):1131-1145. doi: 10.1007/s10545-018-0230-z. Epub 2018 Jul 18. J Inherit Metab Dis. 2018. PMID: 30022305
-
Ketogenic and anaplerotic dietary modifications ameliorate seizure activity in Drosophila models of mitochondrial encephalomyopathy and glycolytic enzymopathy.Mol Genet Metab. 2019 Apr;126(4):439-447. doi: 10.1016/j.ymgme.2019.01.008. Epub 2019 Jan 17. Mol Genet Metab. 2019. PMID: 30683556 Free PMC article.
-
Mass Spectrometry Imaging Reveals Region-Specific Lipid Alterations in the Mouse Brain in Response to Efavirenz Treatment.ACS Pharmacol Transl Sci. 2024 Jul 9;7(8):2379-2390. doi: 10.1021/acsptsci.4c00228. eCollection 2024 Aug 9. ACS Pharmacol Transl Sci. 2024. PMID: 39156742 Free PMC article.
References
-
- Schneider AS. Triosephosphate isomerase deficiency: historical perspectives and molecular aspects. Baillieres Best Pract Res Clin Haematol. 2000;13(1):119–40. Epub 2000/08/05. doi: S1521692600900616 [pii]. . - PubMed
-
- Miwa S, Fujii H. Molecular basis of erythroenzymopathies associated with hereditary hemolytic anemia: tabulation of mutant enzymes. Am J Hematol. 1996;51(2):122–32. Epub 1996/02/01. 10.1002/(SICI)1096-8652(199602)51:2<122::AID-AJH5>3.0.CO;2-# [pii] 10.1002/(SICI)1096-8652(199602)51:2<122::AID-AJH5>3.0.CO;2-#. . - DOI - PubMed
-
- Climent F, Roset F, Repiso A, Perez de la Ossa P. Red cell glycolytic enzyme disorders caused by mutations: an update. Cardiovasc Hematol Disord Drug Targets. 2009;9(2):95–106. Epub 2009/06/13. . - PubMed
-
- Sarper N, Zengin E, Jakobs C, Salomons GS, Mc Wamelink M, Ralser M, et al. Mild hemolytic anemia, progressive neuromotor retardation and fatal outcome: a disorder of glycolysis, triose- phosphate isomerase deficiency. Turk J Pediatr. 2013;55(2):198–202. Epub 2013/11/07. . - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
