

Ultra-Deep Sequencing of HIV-1 near Full-Length and Partial Proviral Genomes Reveals High Genetic Diversity among Brazilian Blood Donors
- PMID: 27031505
- PMCID: PMC4816342
- DOI: 10.1371/journal.pone.0152499
Ultra-Deep Sequencing of HIV-1 near Full-Length and Partial Proviral Genomes Reveals High Genetic Diversity among Brazilian Blood Donors
Abstract
Background: Here, we aimed to gain a comprehensive picture of the HIV-1 diversity in the northeast and southeast part of Brazil. To this end, a high-throughput sequencing-by-synthesis protocol and instrument were used to characterize the near full length (NFLG) and partial HIV-1 proviral genome in 259 HIV-1 infected blood donors at four major blood centers in Brazil: Pro-Sangue foundation (São Paulo state (SP), n 51), Hemominas foundation (Minas Gerais state (MG), n 41), Hemope foundation (Recife state (PE), n 96) and Hemorio blood bank (Rio de Janeiro (RJ), n 70).
Materials and methods: A total of 259 blood samples were obtained from 195 donors with long-standing infections and 64 donors with a lack of stage information. DNA was extracted from the peripheral blood mononuclear cells (PBMCs) to amplify the HIV-1 NFLGs from five overlapping fragments. The amplicons were molecularly bar-coded, pooled, and sequenced by Illumina paired-end protocol.
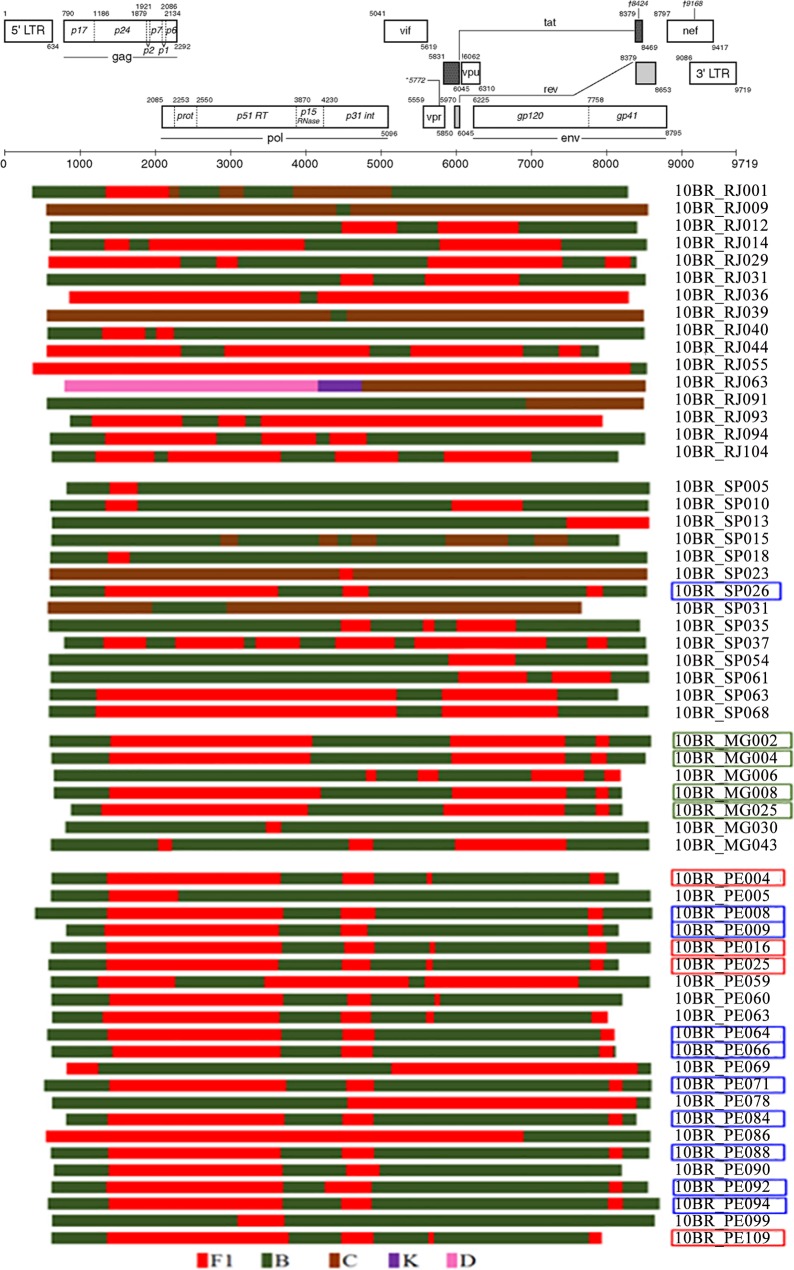
Results: Of the 259 samples studied, 208 (80%) NFLGs and 49 (18.8%) partial fragments were de novo assembled into contiguous sequences and successfully subtyped. Of these 257 samples, 183 (71.2%) were pure subtypes consisting of clade B (n = 167, 65%), C (n = 10, 3.9%), F1 (n = 4, 1.5%), and D (n = 2, 0.7%). Recombinant viruses were detected in 74 (28.8%) samples and consist of unique BF1 (n = 41, 15.9%), BC (n = 7, 2.7%), BCF1 (n = 4, 1.5%), CF1 and CDK (n = 1, 0.4%, each), CRF70_BF1 (n = 4, 1.5%), CRF71_BF1 (n = 12, 4.7%), and CRF72_BF1 (n = 4, 1.5%). Evidence of dual infection was detected in four patients coinfected with the same subtype (n = 3) and distinct subtype (n = 1).
Conclusion: Based on this work, subtype B appears to be the prevalent subtype followed by a high proportion of intersubtype recombinants that appeared to be arising continually in this country. Our study represents the largest analysis of the viral NFLG ever undertaken worldwide and provides insights into the understanding the genesis of the HIV-1 epidemic in this particular area of South America and informs vaccine design and clinical trials.
Conflict of interest statement
Figures



Similar articles
-
Deep sequencing of HIV-1 near full-length proviral genomes identifies high rates of BF1 recombinants including two novel circulating recombinant forms (CRF) 70_BF1 and a disseminating 71_BF1 among blood donors in Pernambuco, Brazil.PLoS One. 2014 Nov 17;9(11):e112674. doi: 10.1371/journal.pone.0112674. eCollection 2014. PLoS One. 2014. PMID: 25401747 Free PMC article.
-
Enhanced detection of viral diversity using partial and near full-length genomes of human immunodeficiency virus Type 1 provirus deep sequencing data from recently infected donors at four blood centers in Brazil.Transfusion. 2015 May;55(5):980-90. doi: 10.1111/trf.12936. Epub 2014 Nov 21. Transfusion. 2015. PMID: 25413141 Free PMC article.
-
Characterization and frequency of a newly identified HIV-1 BF1 intersubtype circulating recombinant form in São Paulo, Brazil.Virol J. 2010 Apr 16;7:74. doi: 10.1186/1743-422X-7-74. Virol J. 2010. PMID: 20398371 Free PMC article.
-
Error rates, PCR recombination, and sampling depth in HIV-1 whole genome deep sequencing.Virus Res. 2017 Jul 15;239:106-114. doi: 10.1016/j.virusres.2016.12.009. Epub 2016 Dec 27. Virus Res. 2017. PMID: 28039047 Review.
-
Transfusion-Acquired HIV: History, Evolution of Screening Tests, and Current Challenges of Unreported Antiretroviral Drug Use in Brazil.Viruses. 2022 Oct 8;14(10):2214. doi: 10.3390/v14102214. Viruses. 2022. PMID: 36298769 Free PMC article. Review.
Cited by
-
HIV-1 genetic diversity and antiretroviral drug resistance among individuals from Roraima state, northern Brazil.PLoS One. 2017 Mar 16;12(3):e0173894. doi: 10.1371/journal.pone.0173894. eCollection 2017. PLoS One. 2017. PMID: 28301548 Free PMC article.
-
Characterization of HIV-1 CRF90_BF1 and putative novel CRFs_BF1 in Central West, North and Northeast Brazilian regions.PLoS One. 2017 Jun 19;12(6):e0178578. doi: 10.1371/journal.pone.0178578. eCollection 2017. PLoS One. 2017. PMID: 28628667 Free PMC article.
-
HIV-1 infection among crack cocaine users in a region far from the epicenter of the HIV epidemic in Brazil: Prevalence and molecular characteristics.PLoS One. 2018 Jul 17;13(7):e0199606. doi: 10.1371/journal.pone.0199606. eCollection 2018. PLoS One. 2018. PMID: 30016324 Free PMC article.
-
Bioinformatic data processing pipelines in support of next-generation sequencing-based HIV drug resistance testing: the Winnipeg Consensus.J Int AIDS Soc. 2018 Oct;21(10):e25193. doi: 10.1002/jia2.25193. J Int AIDS Soc. 2018. PMID: 30350345 Free PMC article.
-
Validation of Variant Assembly Using HAPHPIPE with Next-Generation Sequence Data from Viruses.Viruses. 2020 Jul 14;12(7):758. doi: 10.3390/v12070758. Viruses. 2020. PMID: 32674515 Free PMC article.
References
-
- Robertson DL, Anderson JP, Bradac JA, Carr JK, Foley B, Funkhouser RK, et al. HIV-1 nomenclature proposal. Science. 2000;288(5463):55–6. Epub 2000/04/15. . - PubMed
-
- Essex M. Human immunodeficiency viruses in the developing world. Adv Virus Res. 1999;53:71–88. Epub 1999/12/03. . - PubMed
-
- Kuiken C, Thakallapalli R, Esklid A, de Ronde A. Genetic analysis reveals epidemiologic patterns in the spread of human immunodeficiency virus. Am J Epidemiol. 2000;152(9):814–22. Epub 2000/11/21. . - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
