

Renal cell carcinoma escapes death by p53 depletion through transglutaminase 2-chaperoned autophagy
- PMID: 27031960
- PMCID: PMC4823929
- DOI: 10.1038/cddis.2016.14
Renal cell carcinoma escapes death by p53 depletion through transglutaminase 2-chaperoned autophagy
Abstract
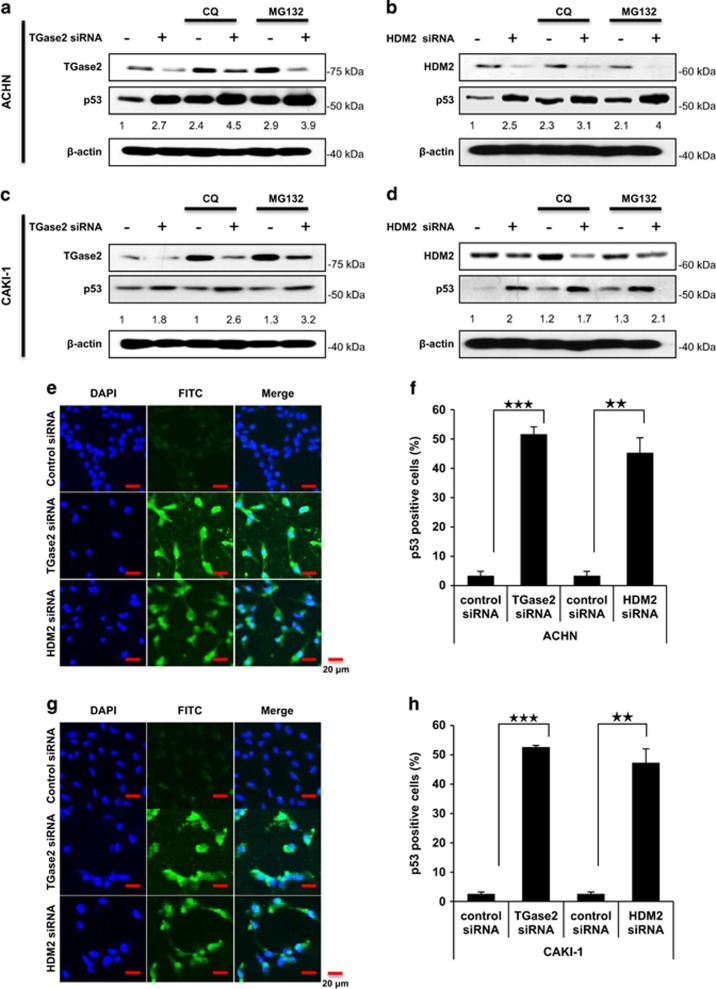
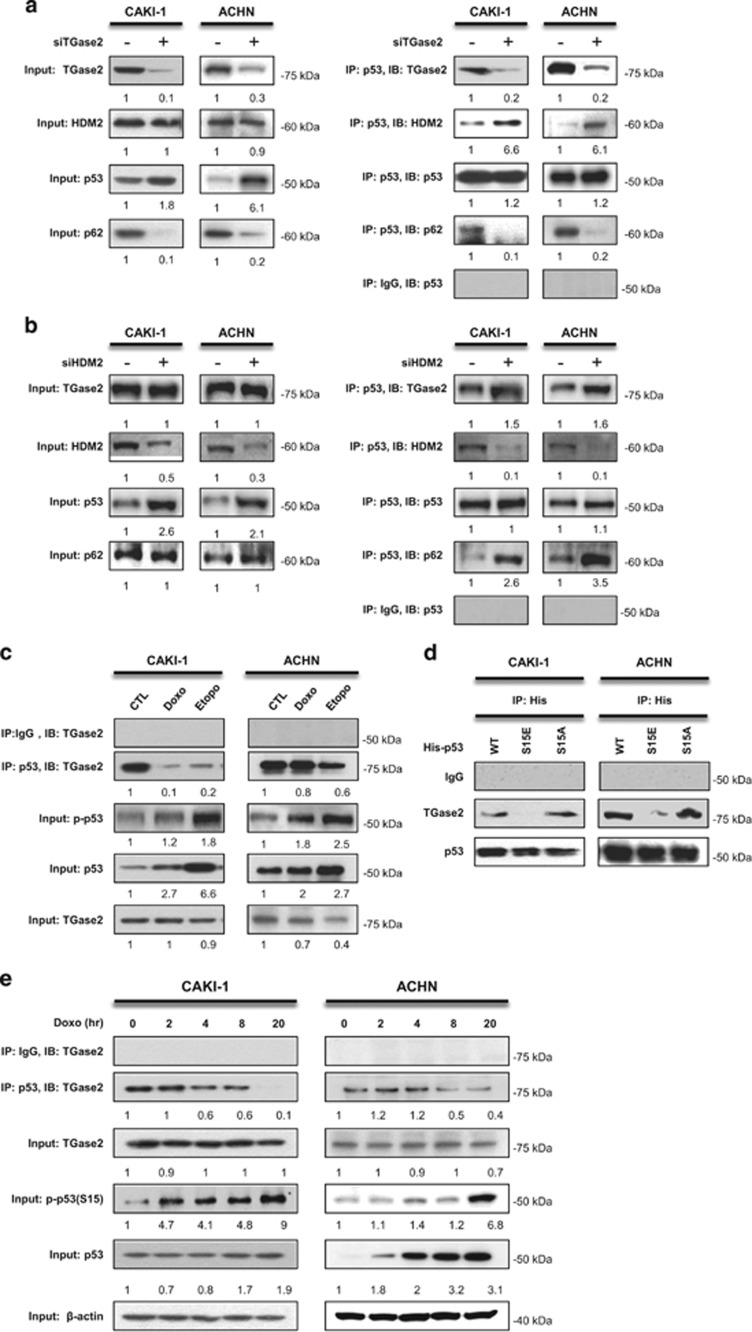
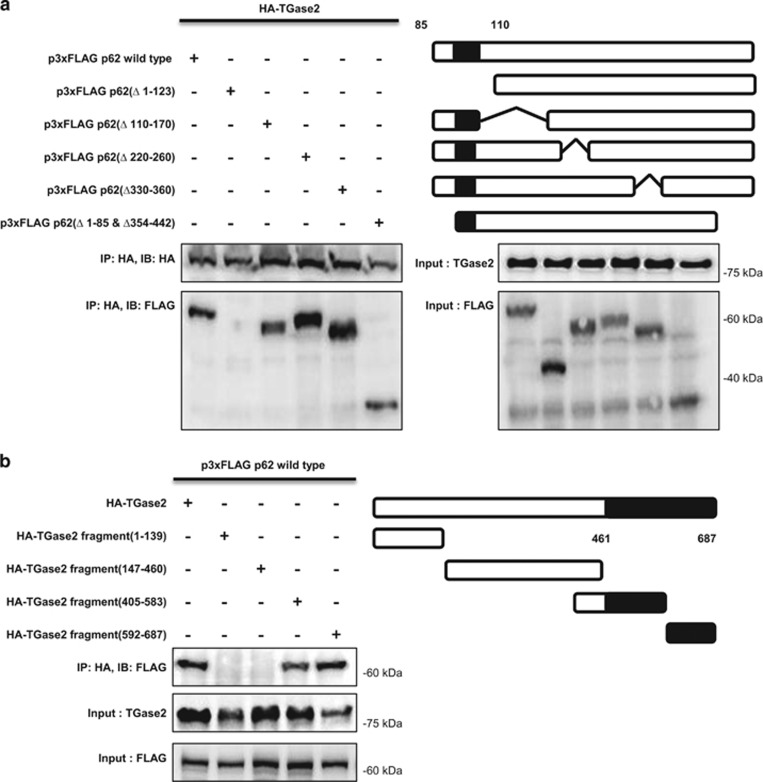
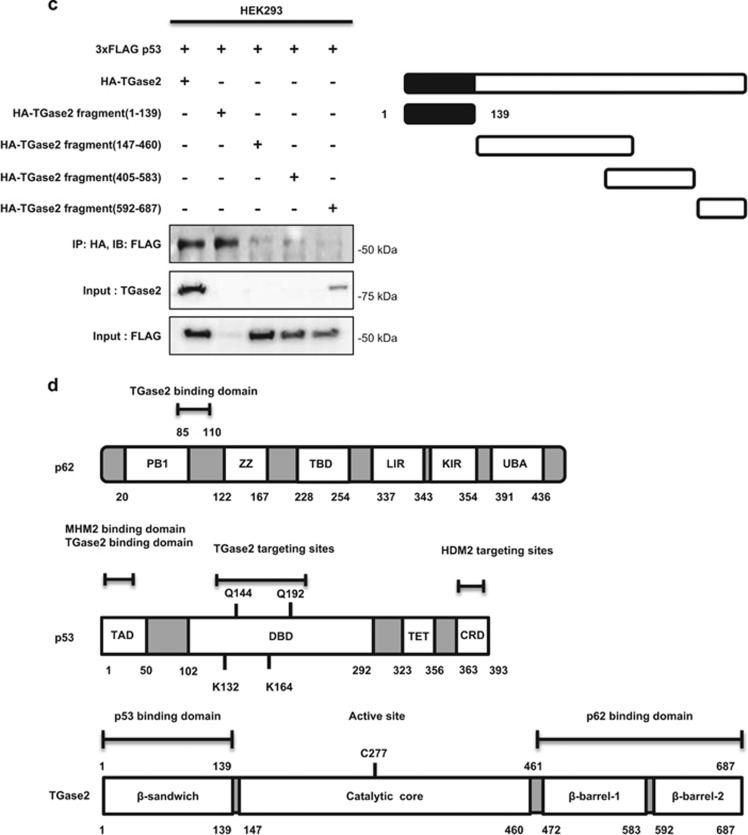
In renal cell carcinoma, transglutaminase 2 (TGase 2) crosslinks p53 in autophagosomes, resulting in p53 depletion and the tumor's evasion of apoptosis. Inhibition of TGase 2 stabilizes p53 and induces tumor cells to enter apoptosis. This study explored the mechanism of TGase 2-dependent p53 degradation. We found that TGase 2 competes with human double minute 2 homolog (HDM2) for binding to p53; promotes autophagy-dependent p53 degradation in renal cell carcinoma (RCC) cell lines under starvation; and binds to p53 and p62 simultaneously without ubiquitin-dependent recognition of p62. The bound complex does not have crosslinking activity. A binding assay using a series of deletion mutants of p62, p53 and TGase 2 revealed that the PB1 (Phox and Bem1p-1) domain of p62 (residues 85-110) directly interacts with the β-barrel domains of TGase 2 (residues 592-687), whereas the HDM2-binding domain (transactivation domain, residues 15-26) of p53 interacts with the N terminus of TGase 2 (residues 1-139). In addition to the increase in p53 stability due to TGase 2 inhibition, the administration of a DNA-damaging anti-cancer drug such as doxorubicin-induced apoptosis in RCC cell lines and synergistically reduced tumor volume in a xenograft model. Combination therapy with a TGase 2 inhibitor and a DNA-damaging agent may represent an effective therapeutic approach for treating RCC.
Figures

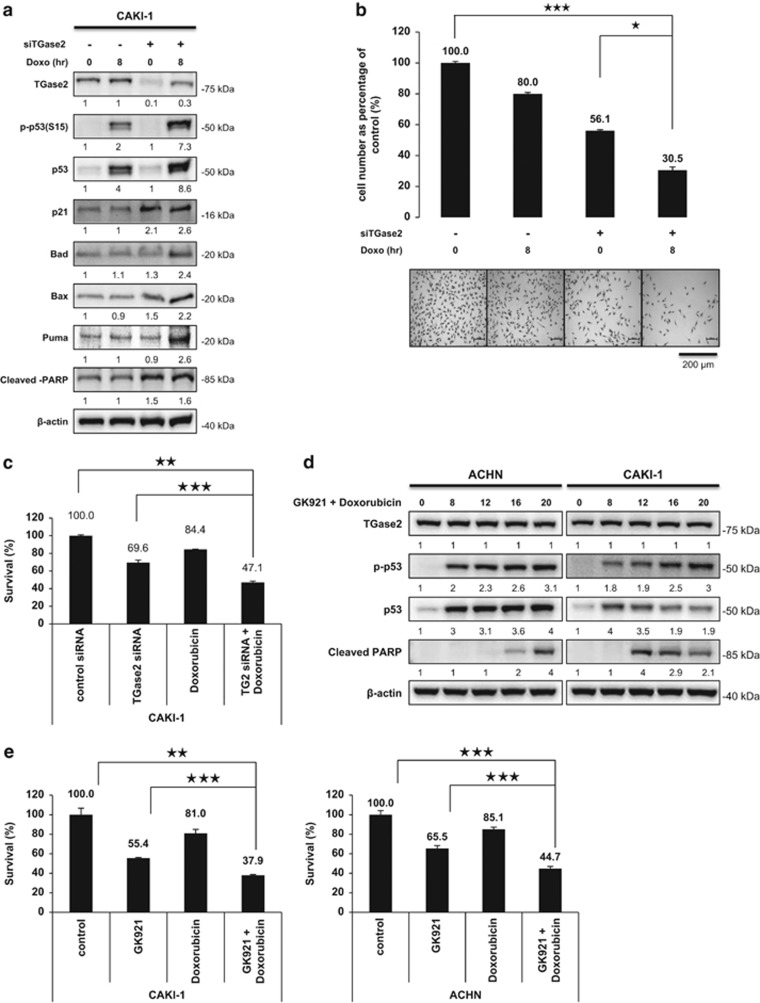
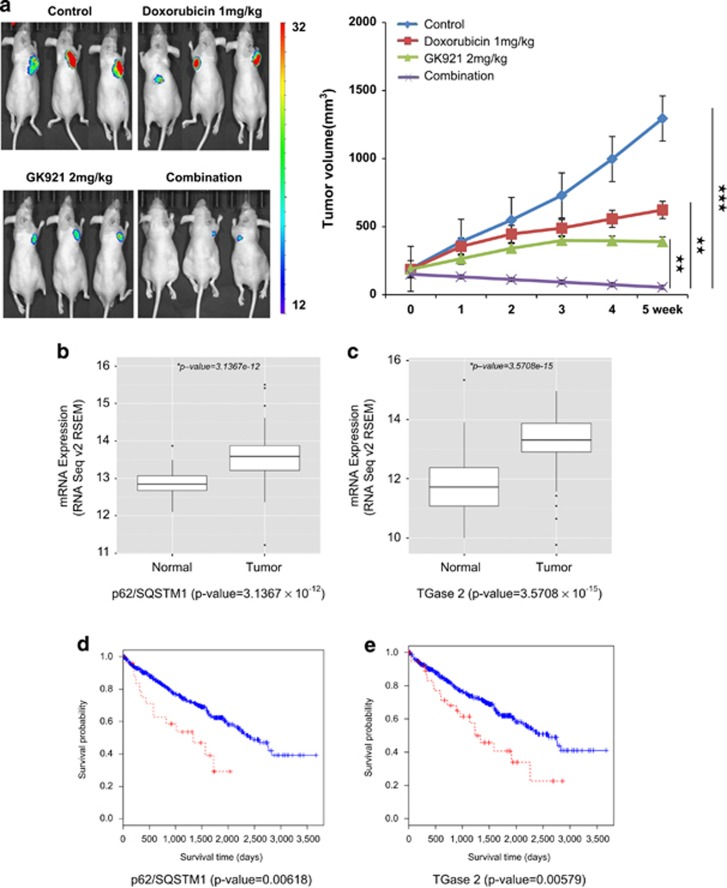
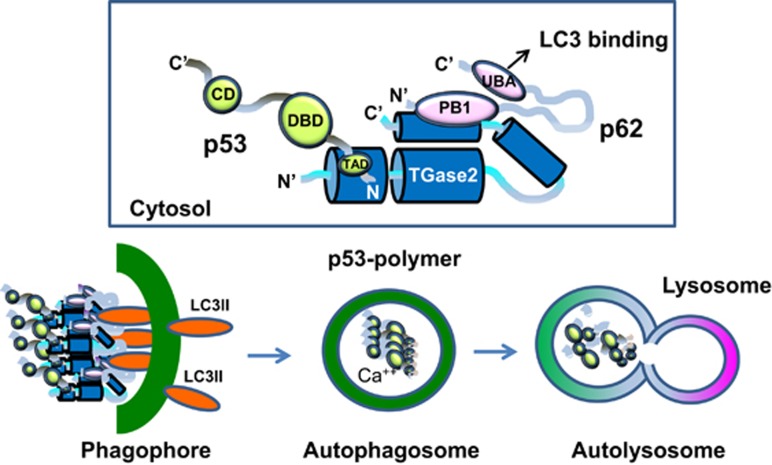
References
-
- Folk JE. Transglutaminases. Annu Rev Biochem 1980; 49: 517–531. - PubMed
-
- Ku BM, Kim DS, Kim KH, Yoo BC, Kim SH, Gong YD et al. Transglutaminase 2 inhibition found to induce p53 mediated apoptosis in renal cell carcinoma. FASEB J 2013; 27: 3487–3495. - PubMed
-
- Oliner JD, Pietenpol JA, Thiagalingam S, Gyuris J, Kinzler KW, Vogelstein B. Oncoprotein MDM2 conceals the activation domain of tumour suppressor p53. Nature 1993; 362: 857–860. - PubMed
-
- Haupt Y, Maya R, Kazaz A, Oren M. Mdm2 promotes the rapid degradation of p53. Nature 1997; 387: 296–299. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
