

p53 downregulates the Fanconi anaemia DNA repair pathway
- PMID: 27033104
- PMCID: PMC4821997
- DOI: 10.1038/ncomms11091
p53 downregulates the Fanconi anaemia DNA repair pathway
Abstract
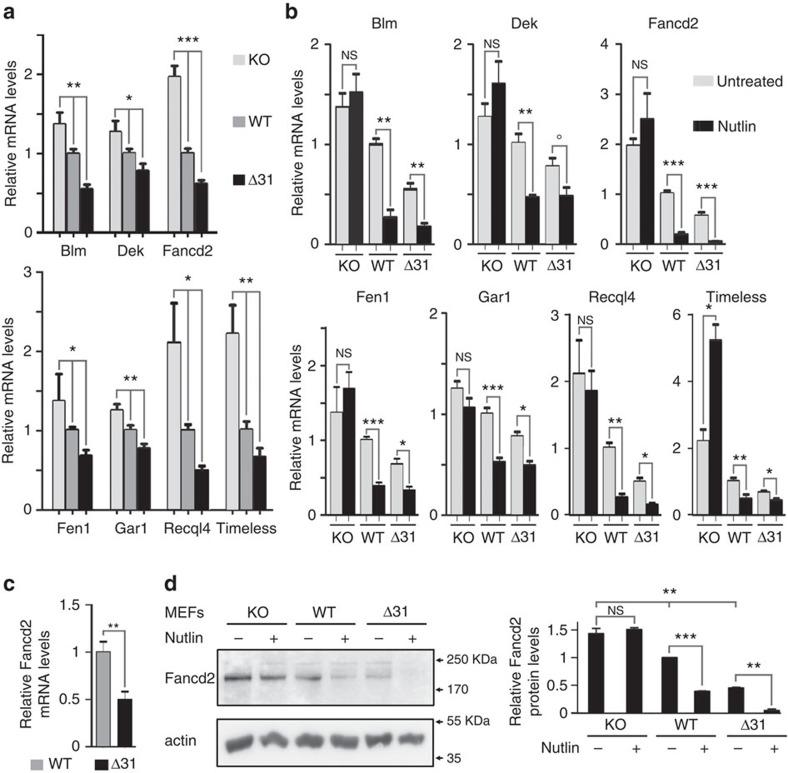
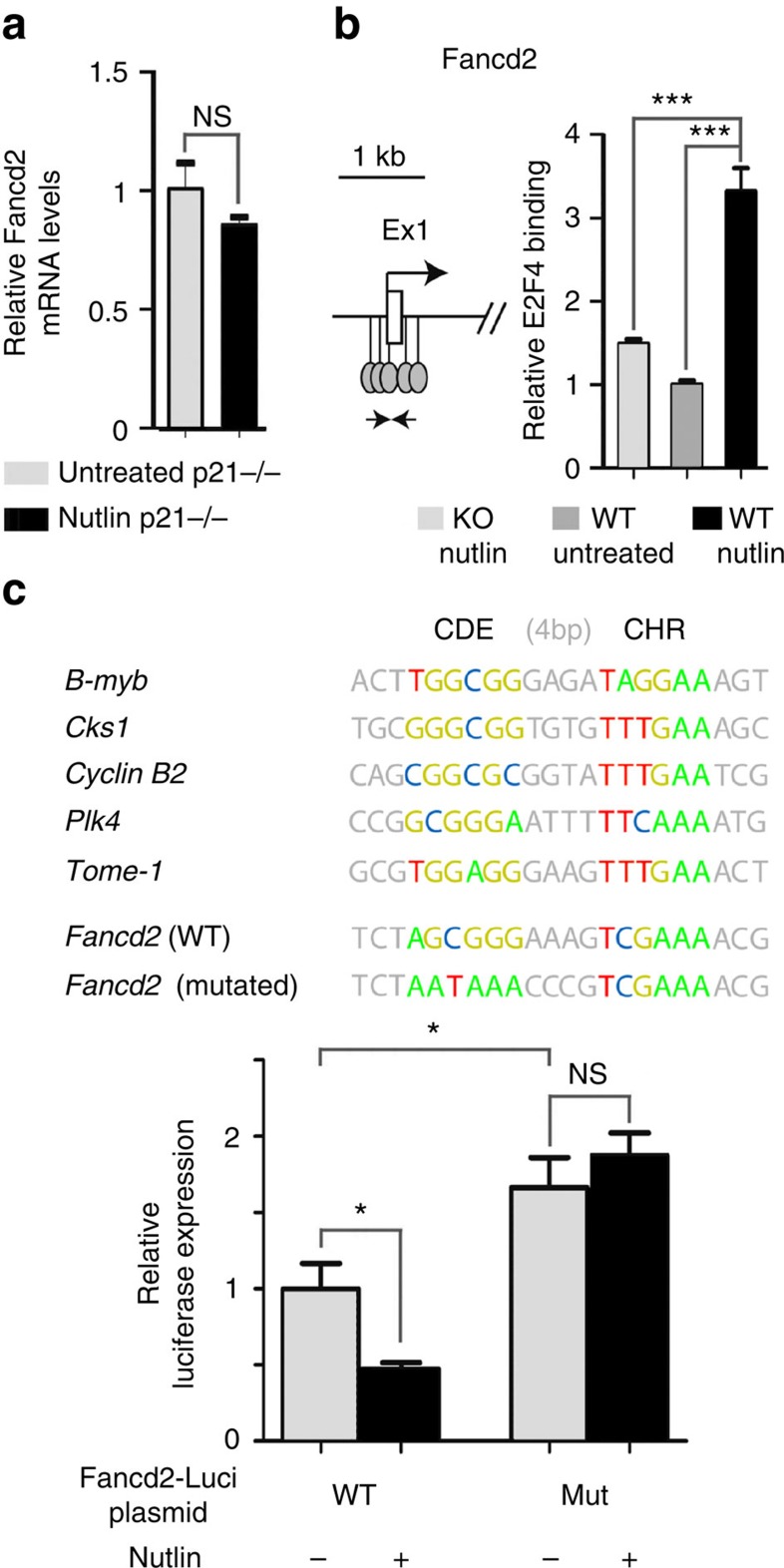
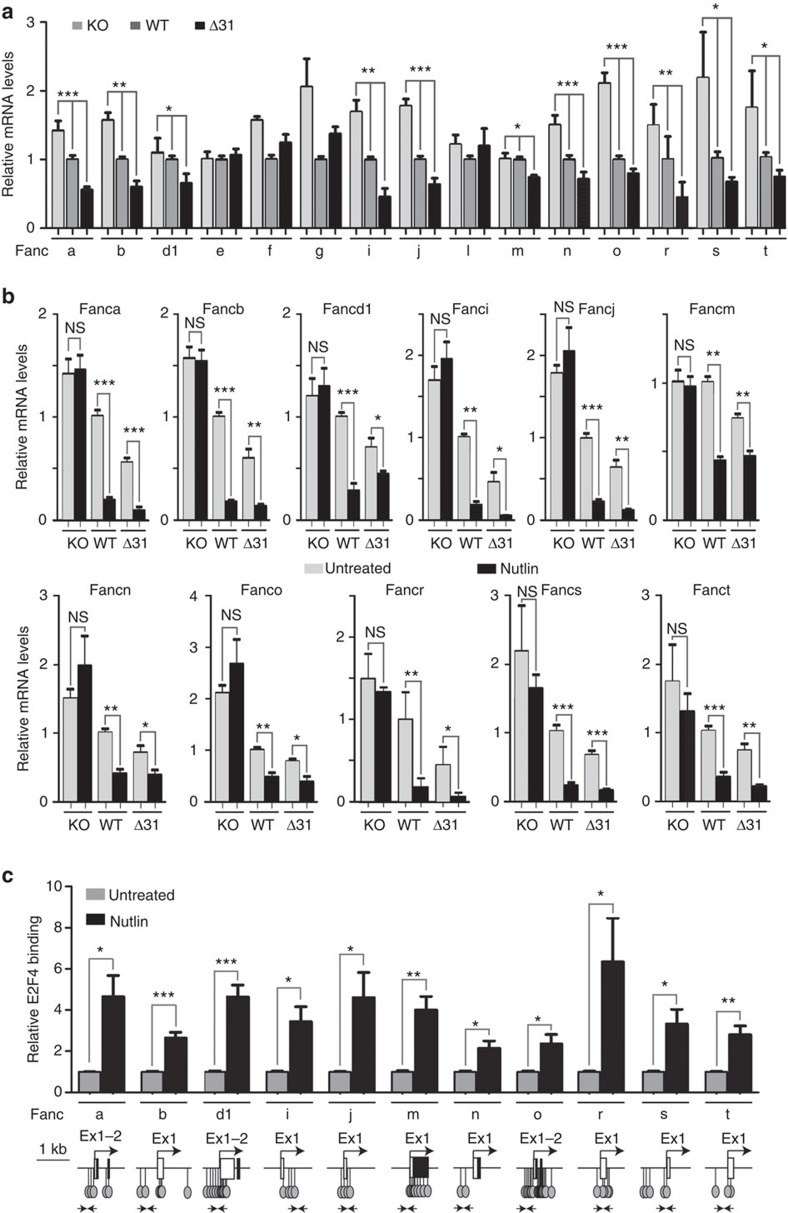
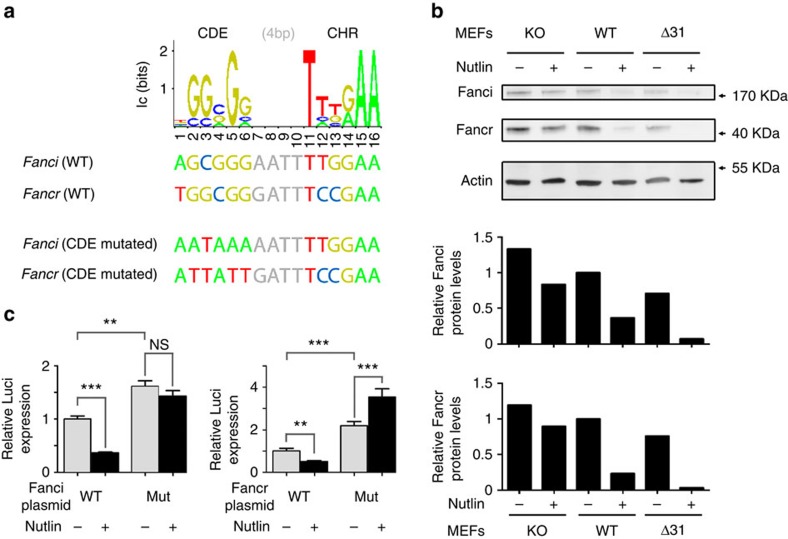
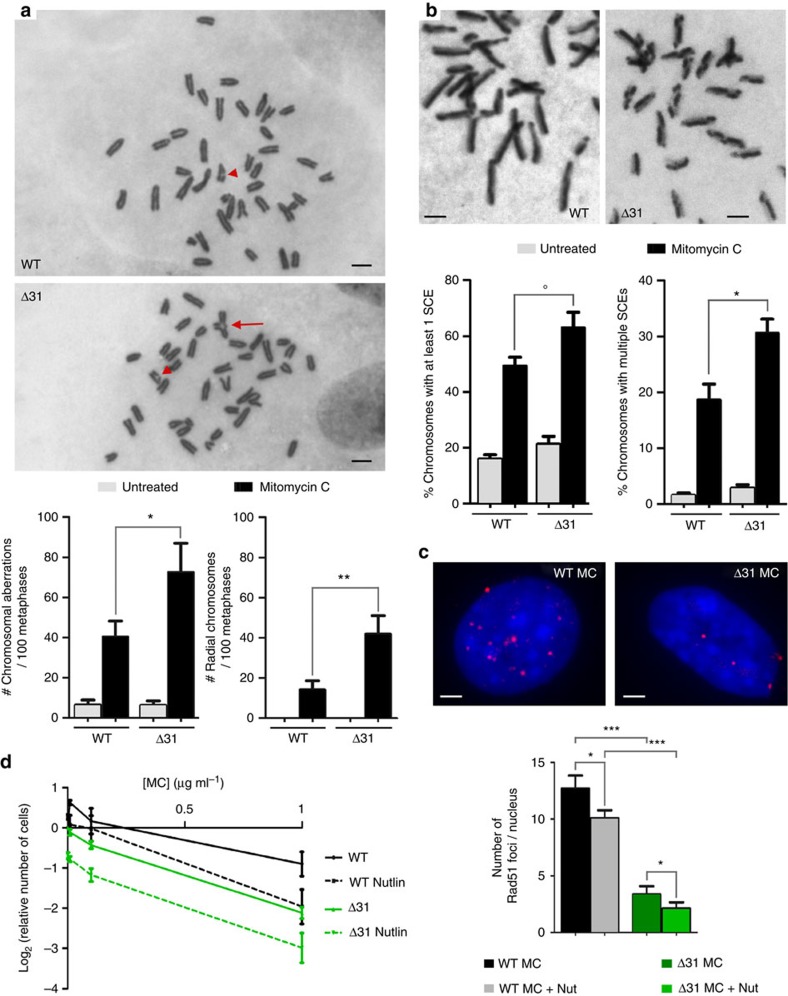
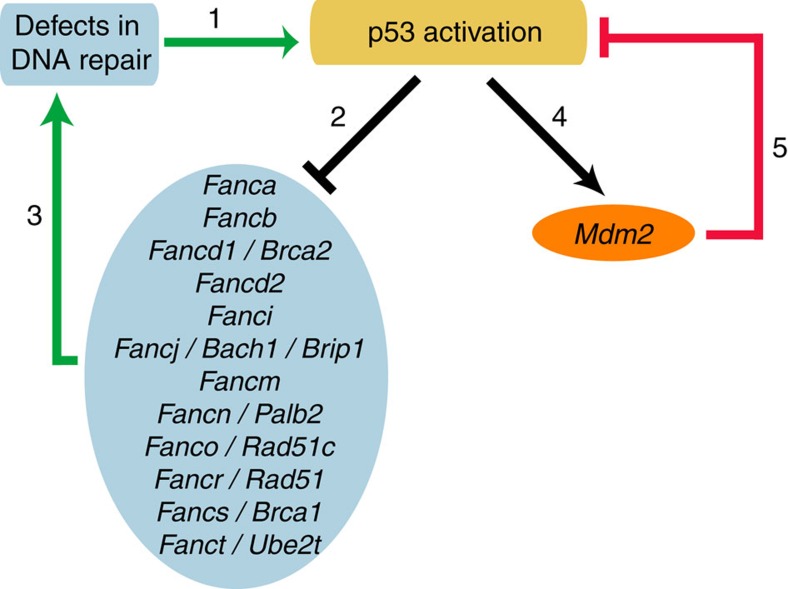
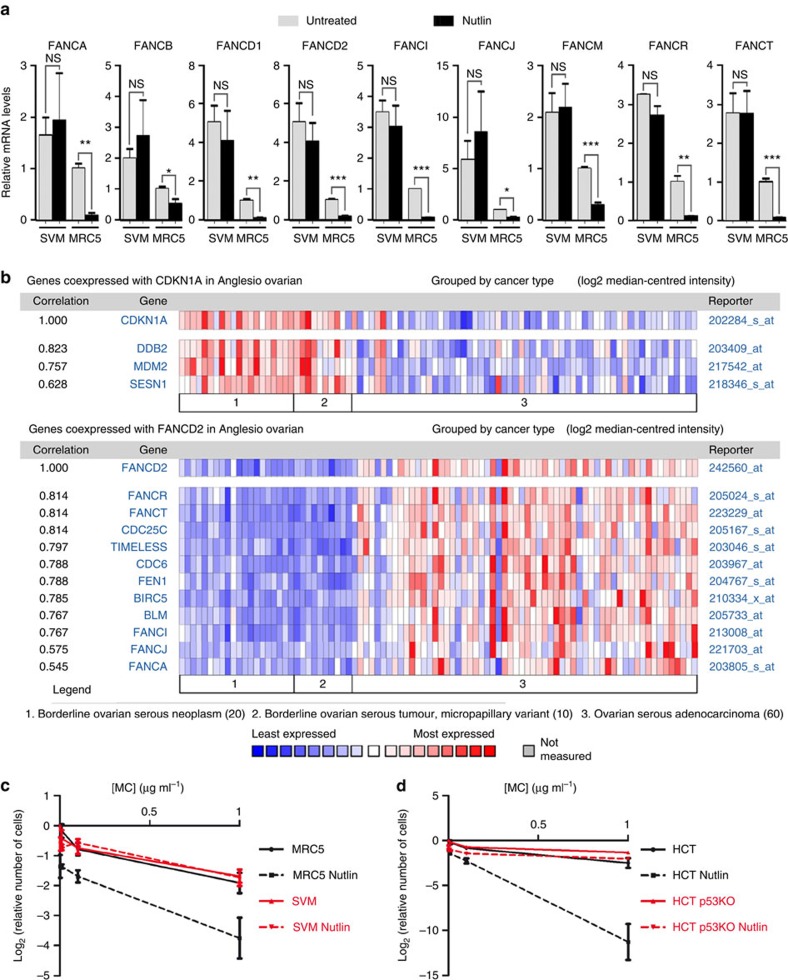
Germline mutations affecting telomere maintenance or DNA repair may, respectively, cause dyskeratosis congenita or Fanconi anaemia, two clinically related bone marrow failure syndromes. Mice expressing p53(Δ31), a mutant p53 lacking the C terminus, model dyskeratosis congenita. Accordingly, the increased p53 activity in p53(Δ31/Δ31) fibroblasts correlated with a decreased expression of 4 genes implicated in telomere syndromes. Here we show that these cells exhibit decreased mRNA levels for additional genes contributing to telomere metabolism, but also, surprisingly, for 12 genes mutated in Fanconi anaemia. Furthermore, p53(Δ31/Δ31) fibroblasts exhibit a reduced capacity to repair DNA interstrand crosslinks, a typical feature of Fanconi anaemia cells. Importantly, the p53-dependent downregulation of Fanc genes is largely conserved in human cells. Defective DNA repair is known to activate p53, but our results indicate that, conversely, an increased p53 activity may attenuate the Fanconi anaemia DNA repair pathway, defining a positive regulatory feedback loop.
Figures
Similar articles
-
[Dangerous liaisons: p53, dyskeratosis congenita and Fanconi anemia].Med Sci (Paris). 2017 Jan;33(1):95-98. doi: 10.1051/medsci/20173301018. Epub 2017 Jan 25. Med Sci (Paris). 2017. PMID: 28120765 French. No abstract available.
-
p53 in the Molecular Circuitry of Bone Marrow Failure Syndromes.Int J Mol Sci. 2023 Oct 6;24(19):14940. doi: 10.3390/ijms241914940. Int J Mol Sci. 2023. PMID: 37834388 Free PMC article. Review.
-
Analysis of a FANCE Splice Isoform in Regard to DNA Repair.J Mol Biol. 2015 Sep 25;427(19):3056-73. doi: 10.1016/j.jmb.2015.08.004. Epub 2015 Aug 12. J Mol Biol. 2015. PMID: 26277624
-
The Fanconi anaemia pathway: new players and new functions.Nat Rev Mol Cell Biol. 2016 Jun;17(6):337-49. doi: 10.1038/nrm.2016.48. Epub 2016 May 5. Nat Rev Mol Cell Biol. 2016. PMID: 27145721 Review.
-
Mutant mice lacking the p53 C-terminal domain model telomere syndromes.Cell Rep. 2013 Jun 27;3(6):2046-58. doi: 10.1016/j.celrep.2013.05.028. Epub 2013 Jun 13. Cell Rep. 2013. PMID: 23770245
Cited by
-
Identifying an AML Prognostic Model Using 10 Marker Genes from Single-Cell Transcriptome and Bulk Transcriptome Analysis.Biochem Genet. 2024 Dec;62(6):4619-4638. doi: 10.1007/s10528-024-10678-9. Epub 2024 Feb 12. Biochem Genet. 2024. PMID: 38347290
-
Cell cycle regulation: p53-p21-RB signaling.Cell Death Differ. 2022 May;29(5):946-960. doi: 10.1038/s41418-022-00988-z. Epub 2022 Mar 31. Cell Death Differ. 2022. PMID: 35361964 Free PMC article. Review.
-
Transcriptional repression of DNA repair genes is a hallmark and a cause of cellular senescence.Cell Death Dis. 2018 Feb 15;9(3):259. doi: 10.1038/s41419-018-0300-z. Cell Death Dis. 2018. PMID: 29449545 Free PMC article.
-
Modern management of Fanconi anemia.Hematology Am Soc Hematol Educ Program. 2022 Dec 9;2022(1):649-657. doi: 10.1182/hematology.2022000393. Hematology Am Soc Hematol Educ Program. 2022. PMID: 36485157 Free PMC article. Review.
-
The Guardian of the Genome Revisited: p53 Downregulates Genes Required for Telomere Maintenance, DNA Repair, and Centromere Structure.Cancers (Basel). 2018 May 6;10(5):135. doi: 10.3390/cancers10050135. Cancers (Basel). 2018. PMID: 29734785 Free PMC article. Review.
References
-
- Simeonova I. et al.. Mutant mice lacking the p53 C-terminal domain model telomere syndromes. Cell Rep. 3, 2046–2058 (2013). - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
