

Review
doi: 10.1038/nrm.2016.27.
Epub 2016 Apr 1.
Non-canonical functions of cell cycle cyclins and cyclin-dependent kinases
Affiliations
- PMID: 27033256
- PMCID: PMC4841706
- DOI: 10.1038/nrm.2016.27
Item in Clipboard
Review
Non-canonical functions of cell cycle cyclins and cyclin-dependent kinases
Nat Rev Mol Cell Biol.
2016 May.
Abstract
The roles of cyclins and their catalytic partners, the cyclin-dependent kinases (CDKs), as core components of the machinery that drives cell cycle progression are well established. Increasing evidence indicates that mammalian cyclins and CDKs also carry out important functions in other cellular processes, such as transcription, DNA damage repair, control of cell death, differentiation, the immune response and metabolism. Some of these non-canonical functions are performed by cyclins or CDKs, independently of their respective cell cycle partners, suggesting that there was a substantial divergence in the functions of these proteins during evolution.
Conflict of interest statement
The authors declare no competing interests.
Figures
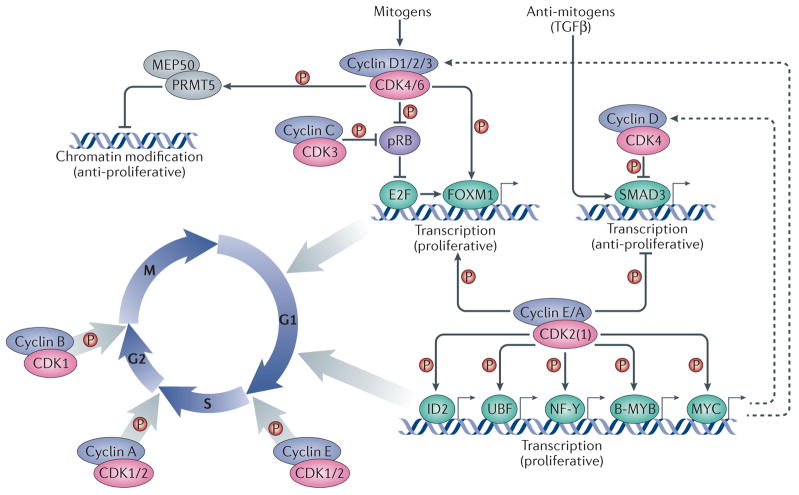
Cell cycle progression is driven by heterodimeric complexes formed by cyclin D, cyclin E, cyclin A or cyclin B with cyclin-dependent kinase (CDK) 4, CDK6, CDK2 or CDK1. (Cyclin C–CDK3 complexes also participate in G0 to G1 phase progression although their physiological relevance in this process is less well established). Cell cycle entry is driven by the formation of cyclin D–CDK4/6 complexes in response to mitogenic stimulation. These complexes phosphorylate and partially inactivate the retinoblastoma protein, pRB, thereby releasing its inhibition of the transcription factor E2F. E2F subsequently promotes the expression of multiple cell cycle genes, among which are E-type cyclins, which bind to and activate CDK2 (and, to a lesser extent, CDK1). Activation of CDK2 (and CDK1) by E-cyclins and A-cyclins leads to the phosphorylation of multiple transcription factors, such as the helix-loop-helix (HLH) protein ID2 as well as UBF, NF-Y, B-MYB and MYC, which contribute to cell cycle progression at different levels. Cyclin D1–CDK4 complexes also phosphorylate chromatin modifiers, such as the protein arginine N-methyltransferase 5 (PRMT5) cofactor MEP50, to promote the repression of specific genes with anti-proliferative properties. Multiple cyclin–CDK complexes can phosphorylate the transcription factors SMAD3, which is activated by antimitogens such as TGFb, and FOXM1 to promote cell cycle progression. Entry into mitosis is specifically driven by cyclin B–CDK1 complexes. Dashed arrows indicate indirect connections or connections with multiple steps. Abbreviations: MEP50, methylosome protein 50; NF-Y, nuclear factor Y; UBF, upstream binding factor.
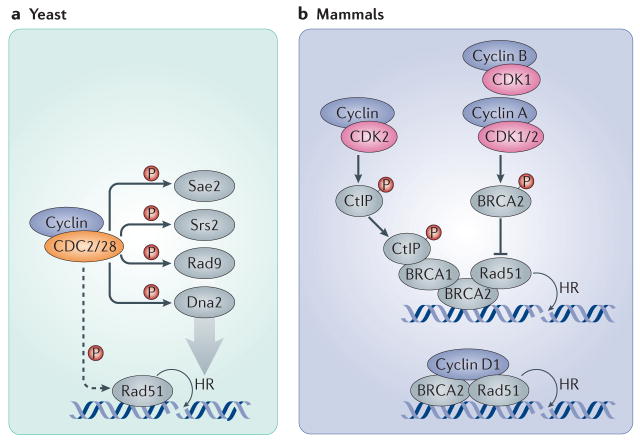
a| In yeast, the sole CDK (CDC28 in S. cerevisiae and CDC2 in S. pombe) participates in the recruitment of Rad51 to DNA double strand breaks and phosphorylates several proteins involved in the DNA damage response and the repair of DSBs through homologous recombination, such as the endonuclease Sae2, the helicase Srs2, the G2 checkpoint protein Rad9 and the resection nuclease Dna2. b| In mammalian cells, CDK2 and CDK1 (together with A-, B- and potentially also E-type cyclins) modulate homologous recombination by the direct phosphorylation of BRCA2, preventing it from binding and recruiting RAD51. Conversely, CDK2 can support homologous recombination by phosphorylating CtBP-interacting protein (CtIP; the human homologue of Sae2), which promotes the recruitment of BRCA1 to DSBs. In addition, cyclin D1 helps to recruit RAD51 to DSB sites by directly interacting with it in a CDK-independent manner, thereby promoting homologous recombination. Dashed arrows indicate indirect connections or connections with multiple steps.
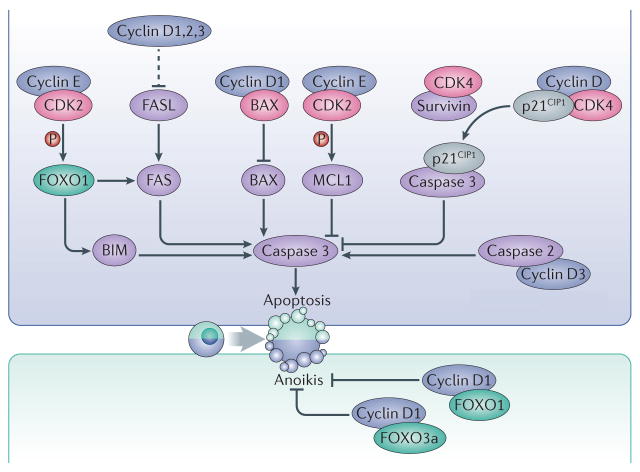
Interphase cyclins modulate apoptosis using CDK-dependent and independent mechanisms. Cyclin E–CDK2 complexes phosphorylate the prosurvival factor MCL1 as well as the transcription factor FOXO1, leading to inhibition or activation of apoptosis, respectively. Phosphorylation of MCL1 stabilizes the protein while phosphorylation of FOXO1 leads to upregulated expression of the pro-apoptotic factors FAS and BIM. Binding of survivin to CDK4 causes the release of the CDK inhibitor p21 (CIP1), which binds and inhibits caspase 3. D-type cyclins can also directly interact with the pro-apoptotic proteins BAX or caspase 2. Moreover, the three D-type cyclins have overlapping roles in repressing the expression of the death receptor FAS and its ligand, FASL, thereby preventing apoptosis in haematopoietic cells. Cyclin D1 also interacts with the transcription factors FOXO1 and FOXO3a and prevents anoikis. Dashed arrows indicate indirect connections or connections with multiple steps.
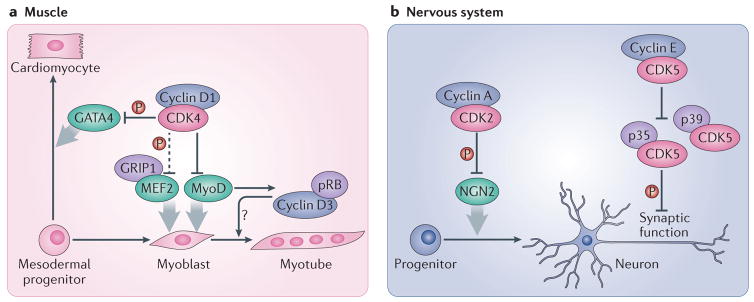
a| In muscle progenitor cells, cyclin D1 interacts with the transcription factor MyoD and inhibits its activity. This effect requires CDK4 but is partially independent of its kinase function. However, the catalytic activity of CDK4 is required for the inhibition by cyclin D1–CDK4 of GRIP1–MEF2 complexes, which are also involved in muscle differentiation. Cyclin D1–CDK4 complexes also phosphorylate and inactivate the GATA4 transcription factor, and thereby repress the differentiation of cardiomyocytes. On the other hand, cyclin D3, which is stabilized in differentiated muscle cells by its interaction with pRB, seems to promote muscle cell differentiation by an unknown mechanism. b| In the nervous system, the differentiation of neuronal progenitors into neurons is blocked by cyclin A–CDK2 complexes, which phosphorylate and inactivate the NGN2 transcription factor. Cyclin E regulates synaptic plasticity by forming kinase-inactive complexes with CDK5 and sequestering it from its activators p35 and p39, thereby inhibiting the phosphorylation of synaptic CDK5 substrates. Dashed arrows indicate indirect connections or connections with multiple steps.
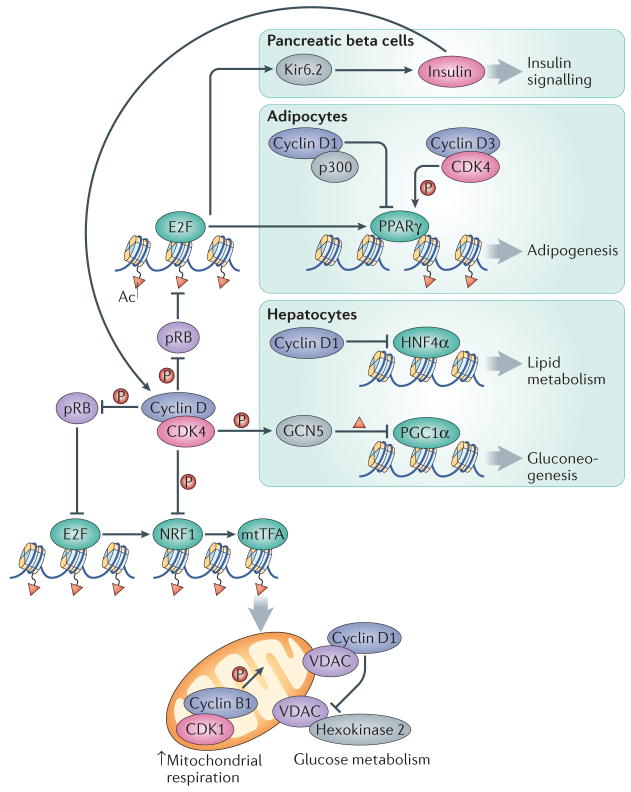
Cyclin–CDK complexes display opposing roles in controlling mitochondrial function. Whereas their activity promotes the expression of the NRF1 factor through the pRB–E2F pathway, cyclin D1-CDK4 can also directly phosphorylate and inactivate NRF1, leading to decreased transcription of mitochondrial proteins by mitochondrial transcription factor A (mtTFA). Moreover, although the activity of mitochondria is directly stimulated by cyclin B1–CDK1, cyclin D1 binding to voltage-dependent anion channel protein (VDAC) prevents hexokinase 2 activation and the use of glucose in mitochondria. In pancreatic beta cells, cyclin D–CDK4 complexes promote insulin secretion through the E2F-mediated expression of the potassium ATP channel component Kir6.2. Insulin, in turn, induces the expression of D-type cyclins through a positive feedback loop. In adipocytes, adipogenesis is inhibited by the interaction of cyclin D1 with the histone acetyltransferase p300, which leads to inhibition of the transcription factor peroxisome proliferator-activated receptor γ (PPARγ). Conversely, adipogenesis is activated by cyclin D3–CDK4 -mediated PPARγ phosphorylation. Gluconeogenesis is inhibited in hepatocytes through the cyclin D1–CDK4-dependent phosphorylation and activation of the acetyltransferase GCN5, which acetylates and inactivates the transcription factor PPARγ coactivator-1α (PGC1α). In addition, cyclin D1 can bind and inactivate the transcription factor hepatocyte nuclear factor-4α (HNF4α), resulting in deficient lipid metabolism.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
