

Mechanisms of FUS mutations in familial amyotrophic lateral sclerosis
- PMID: 27033831
- PMCID: PMC5003642
- DOI: 10.1016/j.brainres.2016.03.036
Mechanisms of FUS mutations in familial amyotrophic lateral sclerosis
Abstract
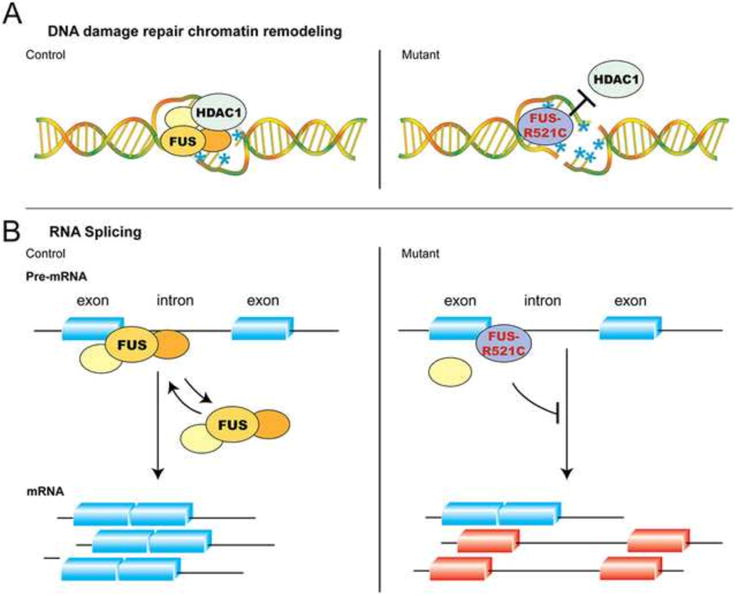
Recent advances in the genetics of amyotrophic lateral sclerosis (ALS) have provided key mechanistic insights to the pathogenesis of this devastating neurodegenerative disease. Among many etiologies for ALS, the identification of mutations and proteinopathies in two RNA binding proteins, TDP-43 (TARDBP or TAR DNA binding protein 43) and its closely related RNA/DNA binding protein FUS (fused in sarcoma), raises the intriguing possibility that perturbations to the RNA homeostasis and metabolism in neurons may contribute to the pathogenesis of these diseases. Although the similarities between TDP-43 and FUS suggest that mutations and proteinopathy involving these two proteins may converge on the same mechanisms leading to neurodegeneration, there is increasing evidence that FUS mutations target distinct mechanisms to cause early disease onset and aggressive progression of disease. This review focuses on the recent advances on the molecular, cellular and genetic approaches to uncover the mechanisms of wild type and mutant FUS proteins during development and in neurodegeneration. These findings provide important insights to understand how FUS mutations may perturb the maintenance of dendrites through fundamental processes in RNA splicing, RNA transport and DNA damage response/repair. These results contribute to the understanding of phenotypic manifestations in neurodegeneration related to FUS mutations, and to identify important directions for future investigations. This article is part of a Special Issue entitled SI:RNA Metabolism in Disease.
Keywords: Amyotrophic lateral sclerosis (ALS); DNA damage repair; Frontotemporal dementia (FTD); Fused in sarcoma (FUS); Low complexity domain; Prion-like property; RNA binding protein; RNA splicing.
Copyright © 2016 The Authors. Published by Elsevier B.V. All rights reserved.
Figures



References
-
- Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, Mann D, Tsuchiya K, Yoshida M, Hashizume Y, et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun. 2006;351:602–611. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
