

The implications of whole-genome sequencing in the control of tuberculosis
- PMID: 27034776
- PMCID: PMC4784569
- DOI: 10.1177/2049936115624630
The implications of whole-genome sequencing in the control of tuberculosis
Abstract
The availability of whole-genome sequencing (WGS) as a tool for the diagnosis and clinical management of tuberculosis (TB) offers considerable promise in the fight against this stubborn epidemic. However, like other new technologies, the best application of WGS remains to be determined, for both conceptual and technical reasons. In this review, we consider the potential value of WGS in the clinical laboratory for the detection of Mycobacterium tuberculosis and the prediction of antibiotic resistance. We also discuss issues pertaining to data generation, interpretation and dissemination, given that WGS has to date been generally performed in research labs where results are not necessarily packaged in a clinician-friendly format. Although WGS is far more accessible now than it was in the past, the transition from a research tool to study TB into a clinical test to manage this disease may require further fine-tuning. Improvements will likely come through iterative efforts that involve both the laboratories ready to move TB into the genomic era and the front-line clinical/public health staff who will be interpreting the results to inform management decisions.
Keywords: Mycobacterium tuberculosis; clinical microbiology; diagnostics; drug resistance; whole-genome sequencing.
Figures
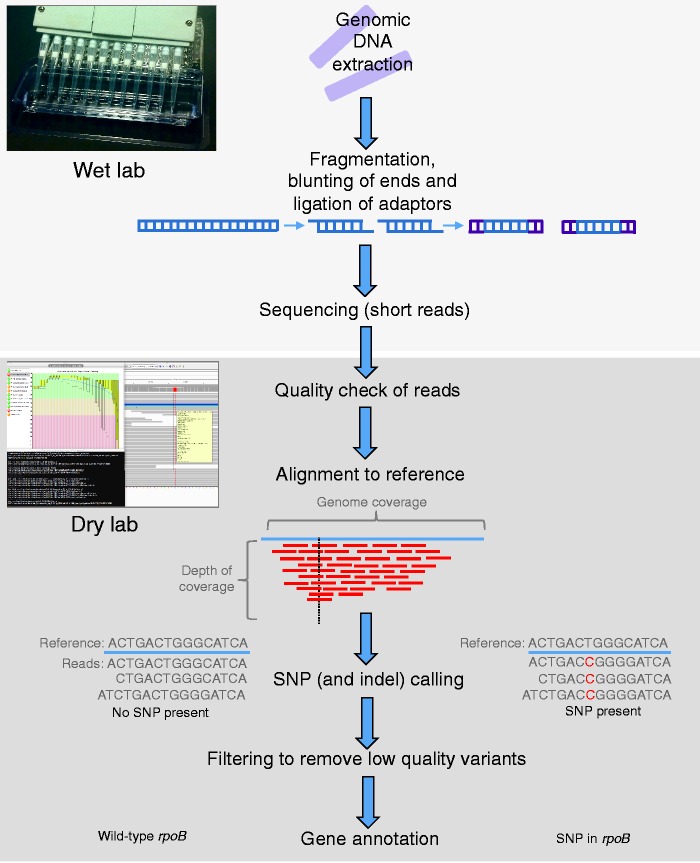
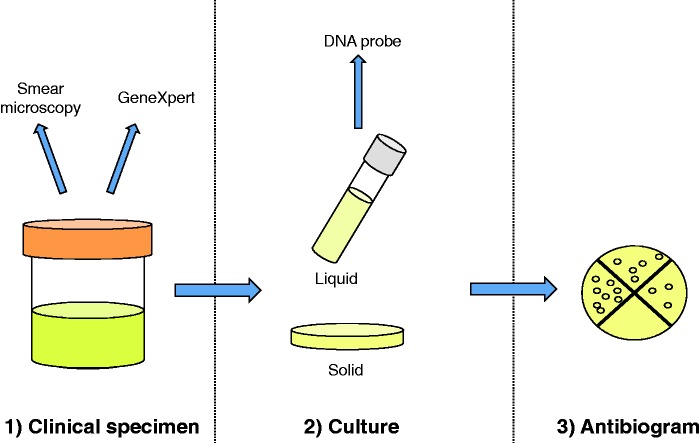
References
-
- Altschul S., Gish W., Miller W., Myers E., Lipman D. (1990) Basic local alignment search tool. J Mol Biol 215: 403–410. - PubMed
-
- Alvarez G., Van Dyk D., Desjardins M., Yasseen A., III, Aaron S., Cameron D., et al. (2015) The feasibility, accuracy, and impact of Xpert MTB/RIF testing in a remote aboriginal community in Canada. Chest 148: 767–773. - PubMed
-
- Behr M., Warren S., Salamon H., Hopewell P., Ponce de Leon A., Daley C., et al. (1999) Transmission of Mycobacterium tuberculosis from patients smear-negative for acid-fast bacilli. Lancet 353: 444–449. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
