

Mutations in chromatin machinery and pediatric high-grade glioma
- PMID: 27034984
- PMCID: PMC4803494
- DOI: 10.1126/sciadv.1501354
Mutations in chromatin machinery and pediatric high-grade glioma
Abstract
Pediatric central nervous system tumors are the most common solid tumor of childhood. Of these, approximately one-third are gliomas that exhibit diverse biological behaviors in the unique context of the developing nervous system. Although low-grade gliomas predominate and have favorable outcomes, up to 20% of pediatric gliomas are high-grade. These tumors are a major contributor to cancer-related morbidity and mortality in infants, children, and adolescents, with long-term survival rates of only 10 to 15%. The recent discovery of somatic oncogenic mutations affecting chromatin regulation in pediatric high-grade glioma has markedly improved our understanding of disease pathogenesis, and these findings have stimulated the development of novel therapeutic approaches targeting epigenetic regulators for disease treatment. We review the current perspective on pediatric high-grade glioma genetics and epigenetics, and discuss the emerging and experimental therapeutics targeting the unique molecular abnormalities present in these deadly childhood brain tumors.
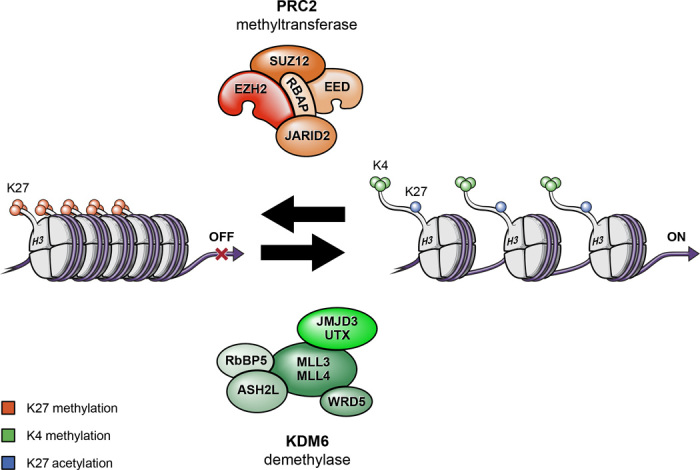
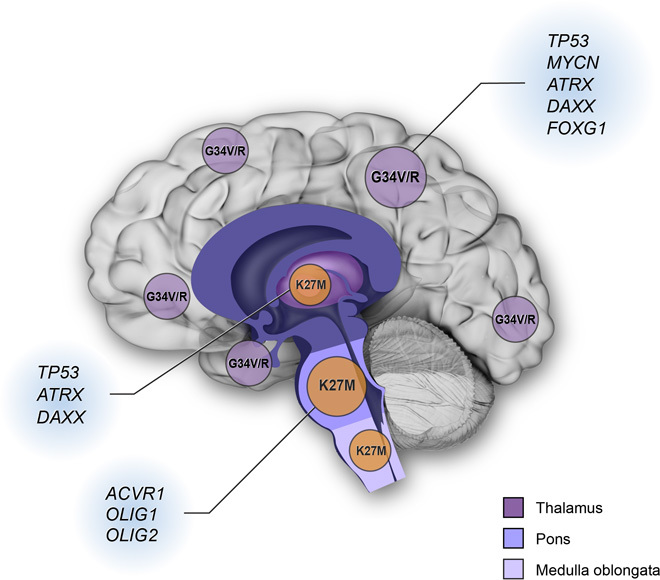
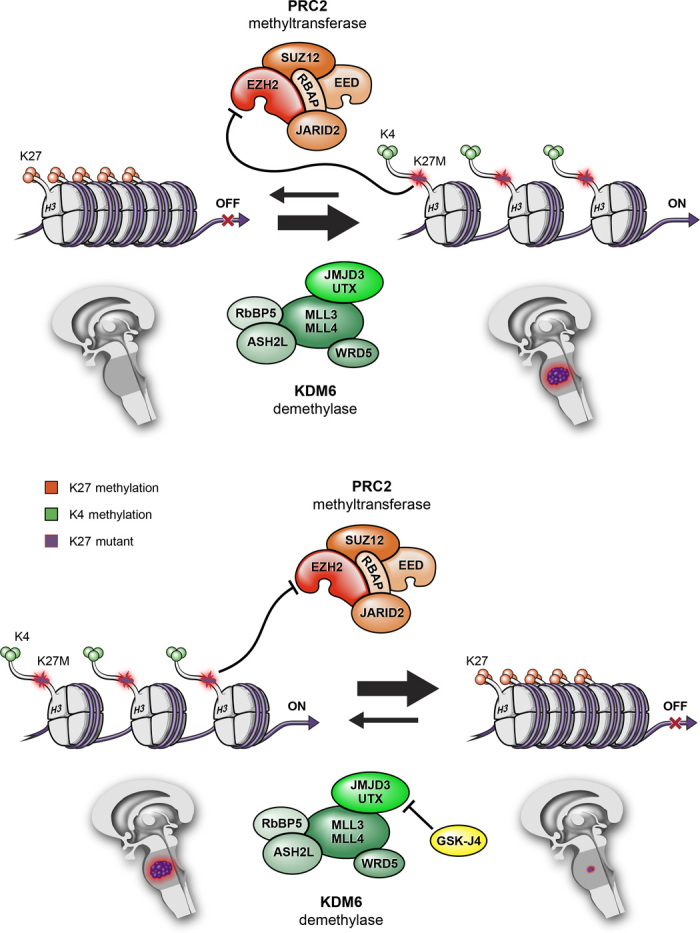
Keywords: DIPG; G34V/R; GSKJ4; Histone mutation; JMJD3; K27M; PRC2; demethylase; methyltransferase; pediatric high-grade glioma.
Figures
References
-
- Sturm D., Witt H., Hovestadt V., Khuong-Quang D.-A., Jones D. T. W., Konermann C., Pfaff E., Tönjes M., Sill M., Bender S., Kool M., Zapatka M., Becker N., Zucknick M., Hielscher T., Liu X.-Y., Fontebasso A. M., Ryzhova M., Albrecht S., Jacob K., Wolter M., Ebinger M., Schuhmann M. U., van Meter T., Frühwald M. C., Hauch H., Pekrun A., Radlwimmer B., Niehues T., von Komorowski G., Dürken M., Kulozik A. E., Madden J., Donson A., Foreman N. K., Drissi R., Fouladi M., Scheurlen W., von Deimling A., Monoranu C., Roggendorf W., Herold-Mende C., Unterberg A., Kramm C. M., Felsberg J., Hartmann C., Wiestler B., Wick W., Milde T., Witt O., Lindroth A. M., Schwartzentruber J., Faury D., Fleming A., Zakrzewska M., Liberski P. P., Zakrzewski K., Hauser P., Garami M., Klekner A., Bognar L., Morrissy S., Cavalli F., Taylor M. D., van Sluis P., Koster J., Versteeg R., Volckmann R., Mikkelsen T., Aldape K., Reifenberger G., Collins V. P., Majewski J., Korshunov A., Lichter P., Plass C., Jabado N., Pfister S. M., Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell 22, 425–437 (2012). - PubMed
-
- Sturm D., Bender S., Jones D. T. W., Lichter P., Grill J., Becher O., Hawkins C., Majewski J., Jones C., Costello J. F., Iavarone A., Aldape K., Brennan C. W., Jabado N., Pfister S. M., Paediatric and adult glioblastoma: Multiform (epi)genomic culprits emerge. Nat. Rev. Cancer 14, 92–107 (2014). - PMC - PubMed
-
- Donaldson S. S., Laningham F., Fisher P. G., Advances toward an understanding of brainstem gliomas. J. Clin. Oncol. 24, 1266–1272 (2006). - PubMed
-
- Robison N. J., Kieran M. W., Diffuse intrinsic pontine glioma: A reassessment. J. Neurooncol. 119, 7–15 (2014). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
