

Monogenic Diabetes: What It Teaches Us on the Common Forms of Type 1 and Type 2 Diabetes
- PMID: 27035557
- PMCID: PMC4890265
- DOI: 10.1210/er.2015-1116
Monogenic Diabetes: What It Teaches Us on the Common Forms of Type 1 and Type 2 Diabetes
Abstract
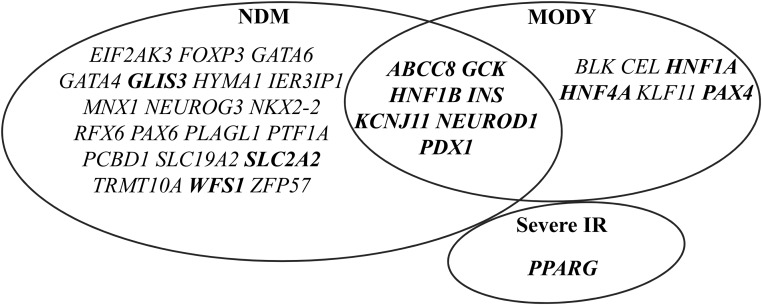
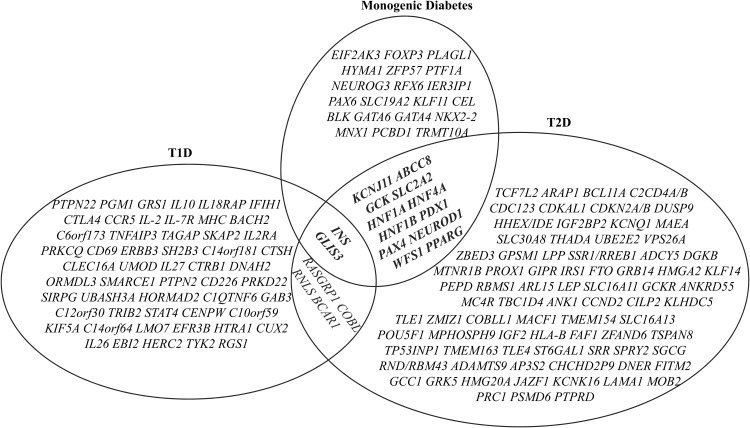
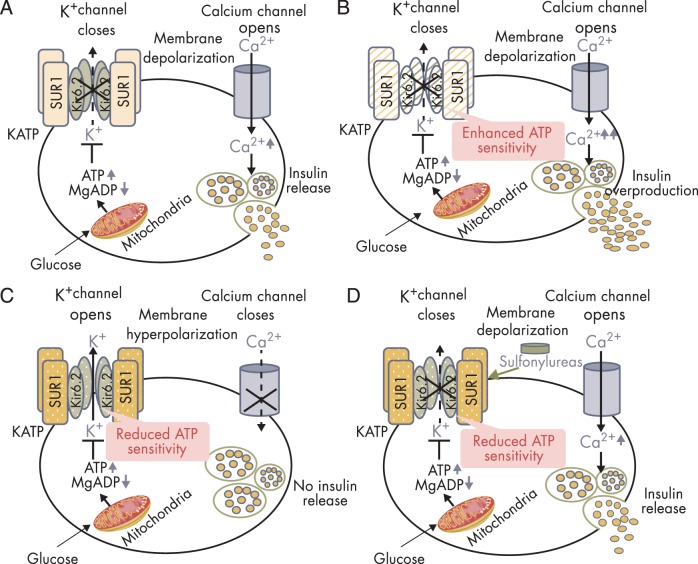
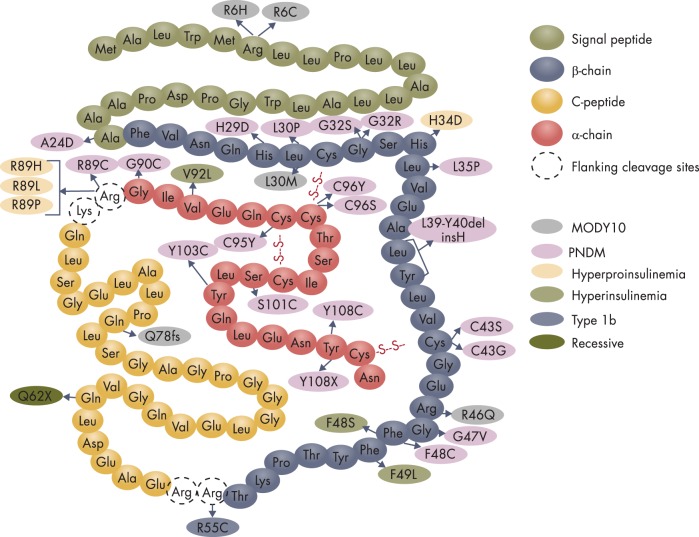
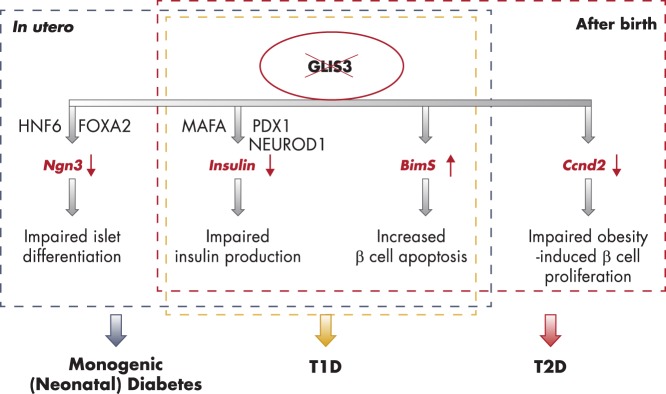
To date, more than 30 genes have been linked to monogenic diabetes. Candidate gene and genome-wide association studies have identified > 50 susceptibility loci for common type 1 diabetes (T1D) and approximately 100 susceptibility loci for type 2 diabetes (T2D). About 1-5% of all cases of diabetes result from single-gene mutations and are called monogenic diabetes. Here, we review the pathophysiological basis of the role of monogenic diabetes genes that have also been found to be associated with common T1D and/or T2D. Variants of approximately one-third of monogenic diabetes genes are associated with T2D, but not T1D. Two of the T2D-associated monogenic diabetes genes-potassium inward-rectifying channel, subfamily J, member 11 (KCNJ11), which controls glucose-stimulated insulin secretion in the β-cell; and peroxisome proliferator-activated receptor γ (PPARG), which impacts multiple tissue targets in relation to inflammation and insulin sensitivity-have been developed as major antidiabetic drug targets. Another monogenic diabetes gene, the preproinsulin gene (INS), is unique in that INS mutations can cause hyperinsulinemia, hyperproinsulinemia, neonatal diabetes mellitus, one type of maturity-onset diabetes of the young (MODY10), and autoantibody-negative T1D. Dominant heterozygous INS mutations are the second most common cause of permanent neonatal diabetes. Moreover, INS gene variants are strongly associated with common T1D (type 1a), but inconsistently with T2D. Variants of the monogenic diabetes gene Gli-similar 3 (GLIS3) are associated with both T1D and T2D. GLIS3 is a key transcription factor in insulin production and β-cell differentiation during embryonic development, which perturbation forms the basis of monogenic diabetes as well as its association with T1D. GLIS3 is also required for compensatory β-cell proliferation in adults; impairment of this function predisposes to T2D. Thus, monogenic forms of diabetes are invaluable "human models" that have contributed to our understanding of the pathophysiological basis of common T1D and T2D.
Figures
References
-
- Steck AK, Winter WE. Review on monogenic diabetes. Curr Opin Endocrinol Diabetes Obes. 2011;18(4):252–258. - PubMed
-
- Edghill EL, Dix RJ, Flanagan SE, et al. HLA genotyping supports a nonautoimmune etiology in patients diagnosed with diabetes under the age of 6 months. Diabetes. 2006;55(6):1895–1898. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
