

Genome-wide kinetics of DNA excision repair in relation to chromatin state and mutagenesis
- PMID: 27036006
- PMCID: PMC4839430
- DOI: 10.1073/pnas.1603388113
Genome-wide kinetics of DNA excision repair in relation to chromatin state and mutagenesis
Abstract
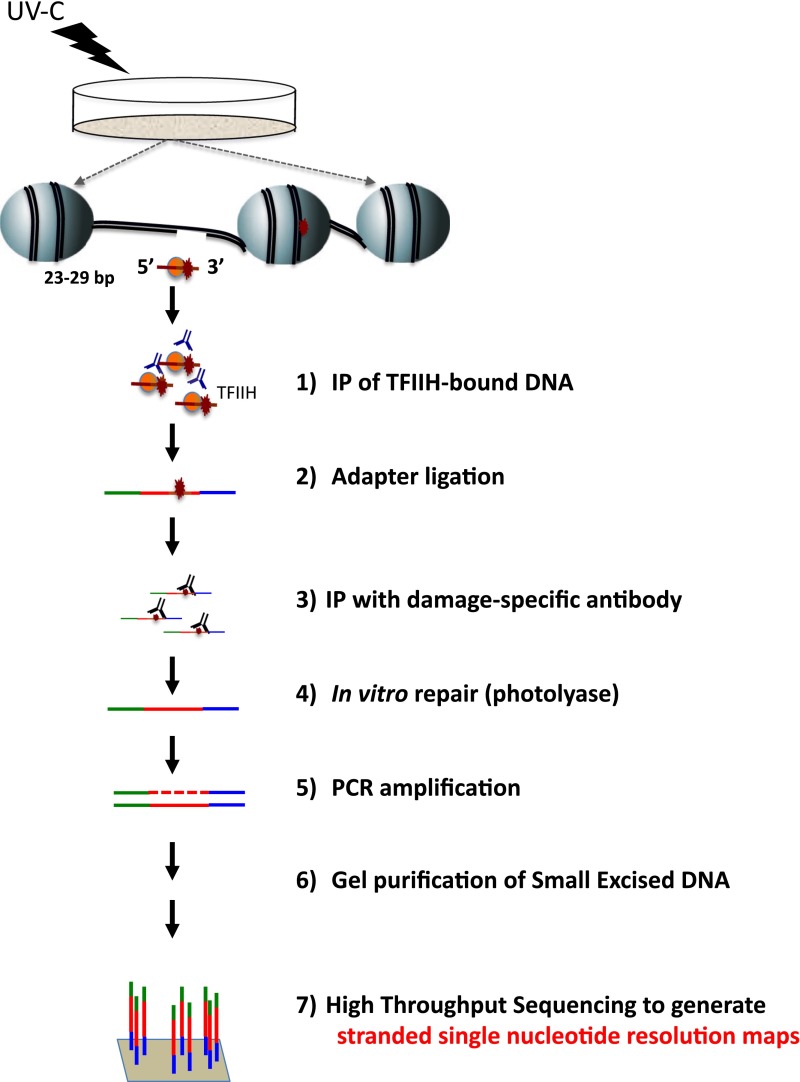
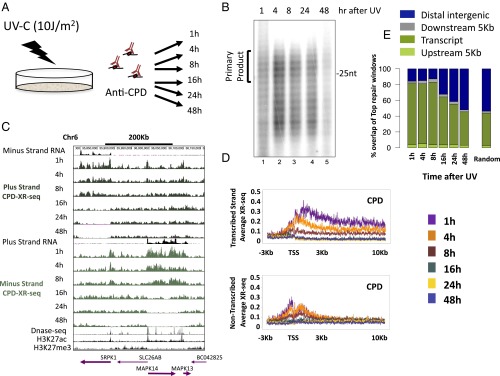
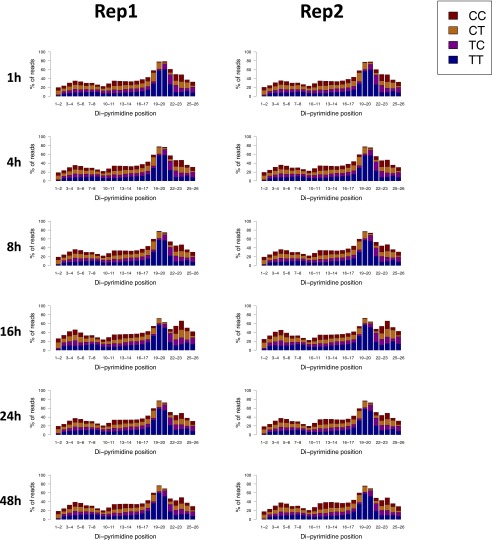
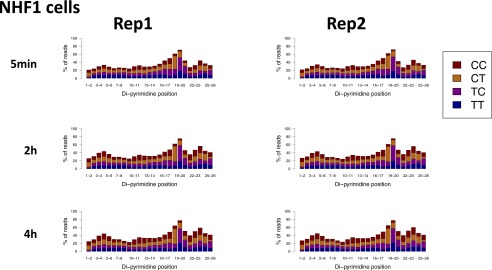
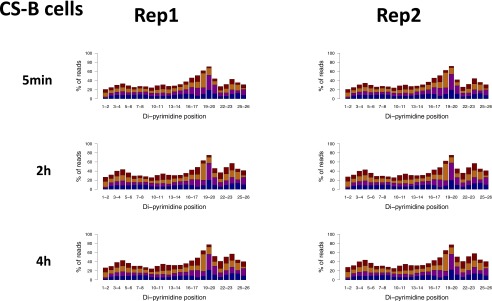
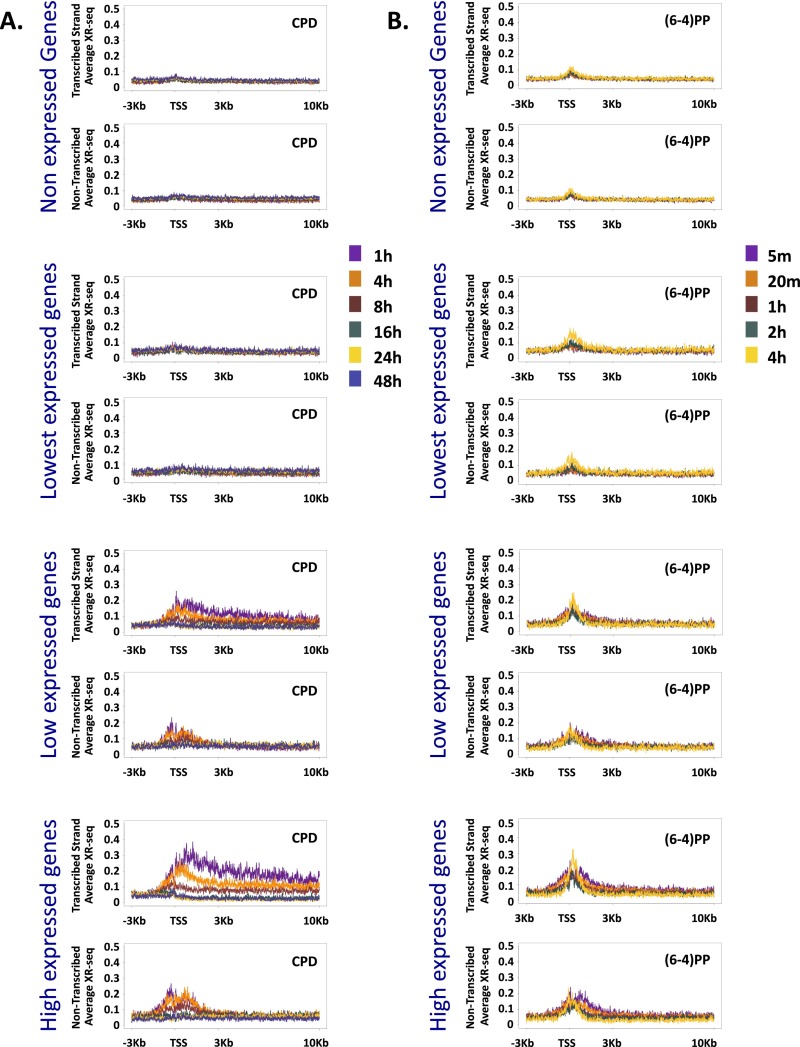
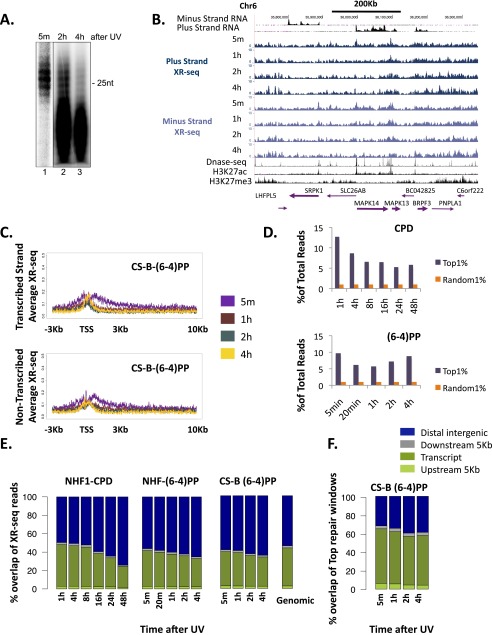
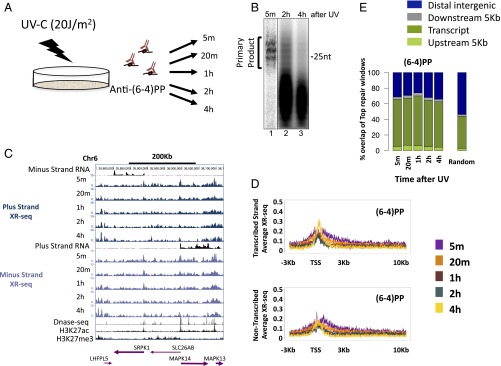
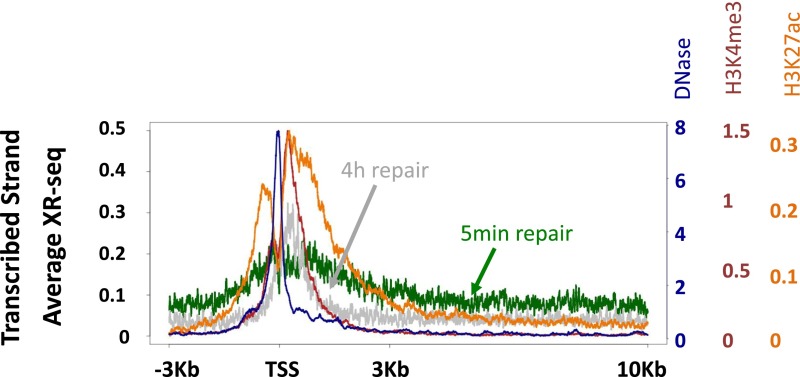
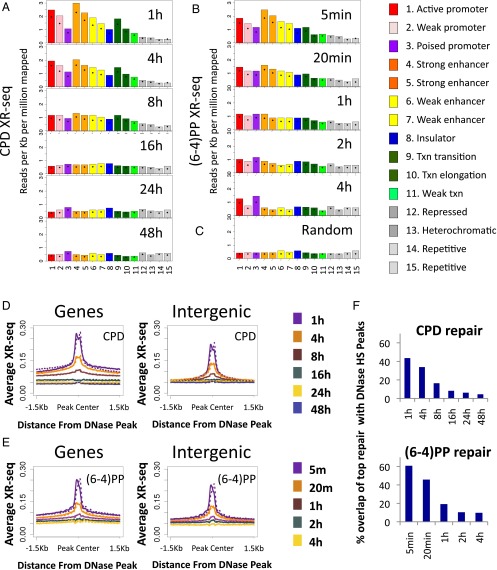
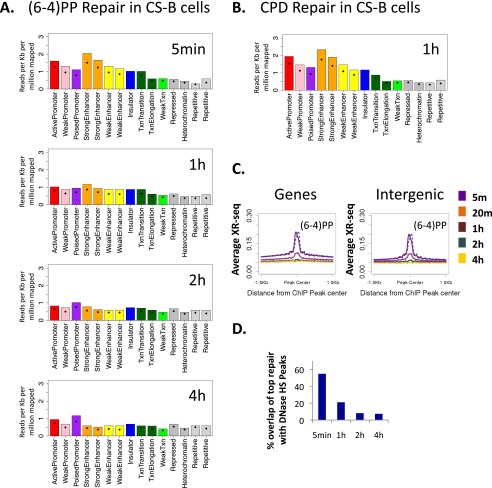
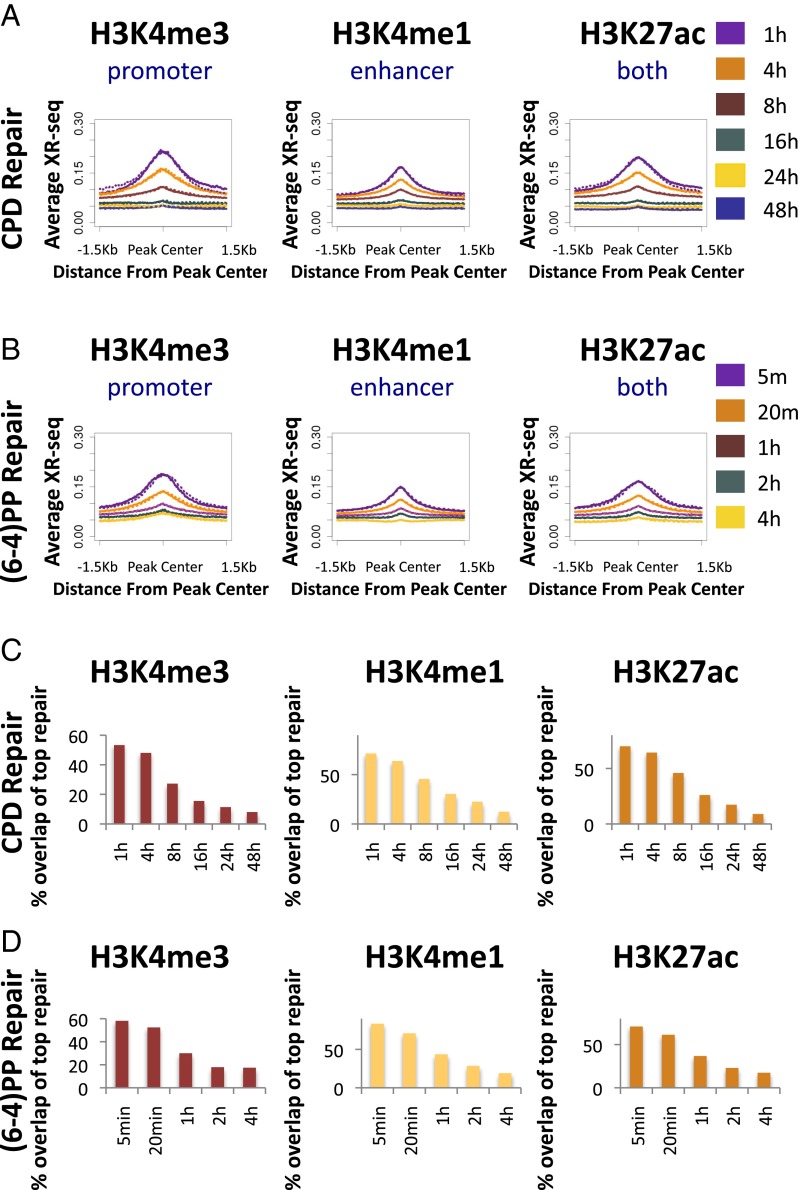
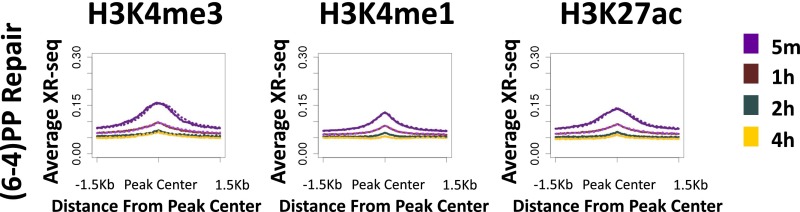
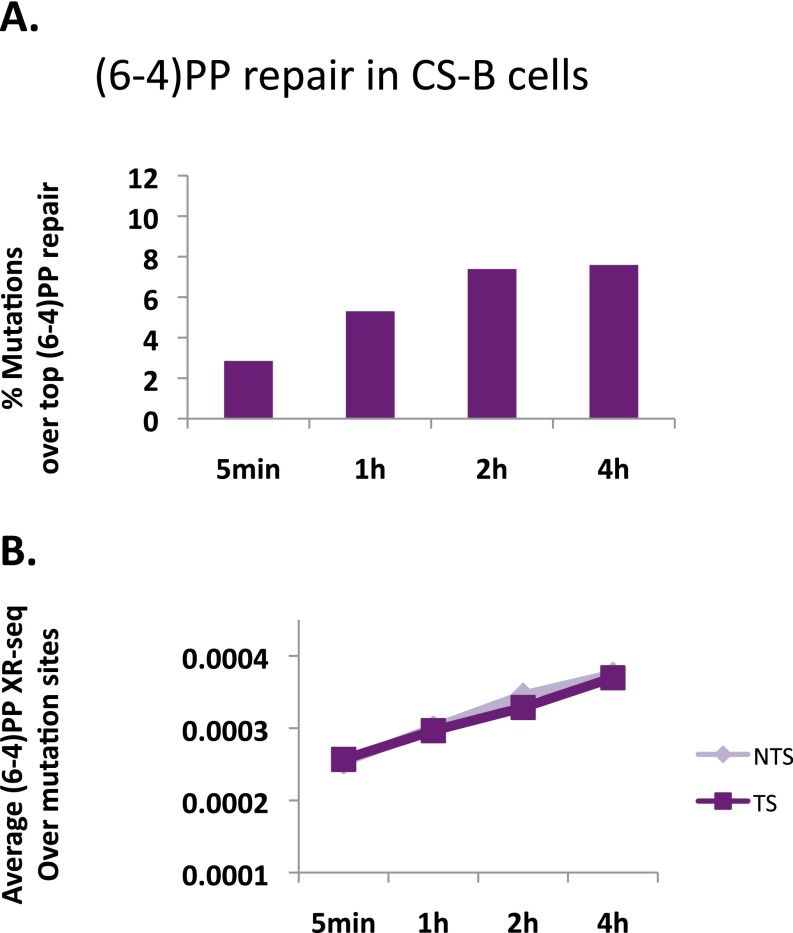
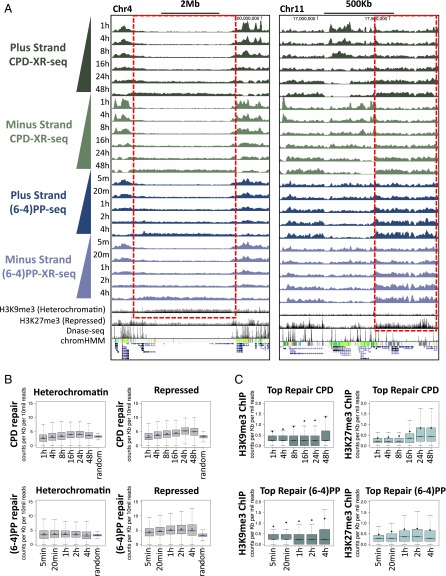
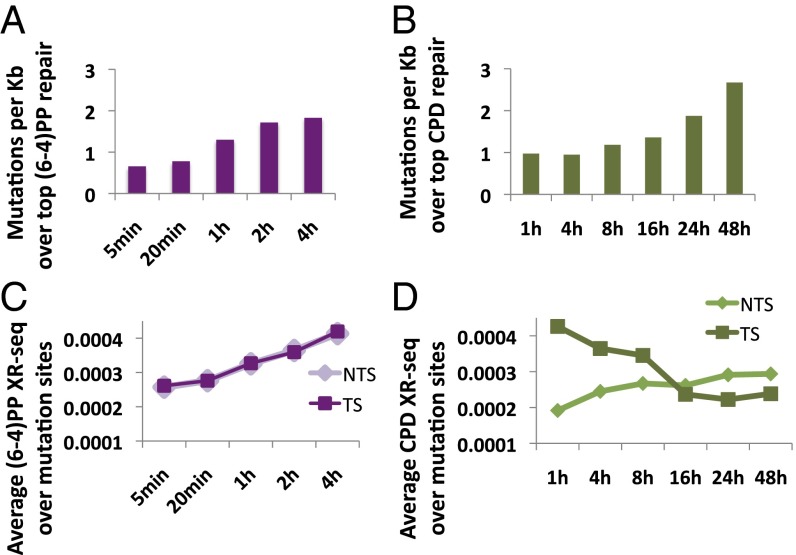
We recently developed a high-resolution genome-wide assay for mapping DNA excision repair named eXcision Repair-sequencing (XR-seq) and have now used XR-seq to determine which regions of the genome are subject to repair very soon after UV exposure and which regions are repaired later. Over a time course, we measured repair of the UV-induced damage of cyclobutane pyrimidine dimers (CPDs) (at 1, 4, 8, 16, 24, and 48 h) and (6-4)pyrimidine-pyrimidone photoproducts [(6-4)PPs] (at 5 and 20 min and 1, 2, and 4 h) in normal human skin fibroblasts. Each type of damage has distinct repair kinetics. The (6-4)PPs are detected as early as 5 min after UV treatment, with the bulk of repair completed by 4 h. Repair of CPDs, which we previously showed is intimately coupled to transcription, is slower and in certain regions persists even 2 d after UV irradiation. We compared our results to the Encyclopedia of DNA Elements data regarding histone modifications, chromatin state, and transcription. For both damage types, and for both transcription-coupled and general excision repair, the earliest repair occurred preferentially in active and open chromatin states. Conversely, repair in regions classified as "heterochromatic" and "repressed" was relatively low at early time points, with repair persisting into the late time points. Damage that remains during DNA replication increases the risk for mutagenesis. Indeed, late-repaired regions are associated with a higher level of cancer-linked mutations. In summary, we show that XR-seq is a powerful approach for studying relationships among chromatin state, DNA repair, genome stability, mutagenesis, and carcinogenesis.
Keywords: DNA damage; DNA repair; chromatin; mutation; transcription.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Reardon JT, Sancar A. Nucleotide excision repair. Prog Nucleic Acid Res Mol Biol. 2005;79(79):183–235. - PubMed
-
- Sancar A. DNA excision repair. Annu Rev Biochem. 1996;65:43–81. - PubMed
-
- Wood RD. Nucleotide excision repair in mammalian cells. J Biol Chem. 1997;272(38):23465–23468. - PubMed
-
- Sugasawa K, et al. Xeroderma pigmentosum group C protein complex is the initiator of global genome nucleotide excision repair. Mol Cell. 1998;2(2):223–232. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
