

A streamlined tethered chromosome conformation capture protocol
- PMID: 27036078
- PMCID: PMC4818521
- DOI: 10.1186/s12864-016-2596-3
A streamlined tethered chromosome conformation capture protocol
Abstract
Background: Identification of locus-locus contacts at the chromatin level provides a valuable foundation for understanding of nuclear architecture and function and a valuable tool for inferring long-range linkage relationships. As one approach to this, chromatin conformation capture-based techniques allow creation of genome spatial organization maps. While such approaches have been available for some time, methodological advances will be of considerable use in minimizing both time and input material required for successful application.
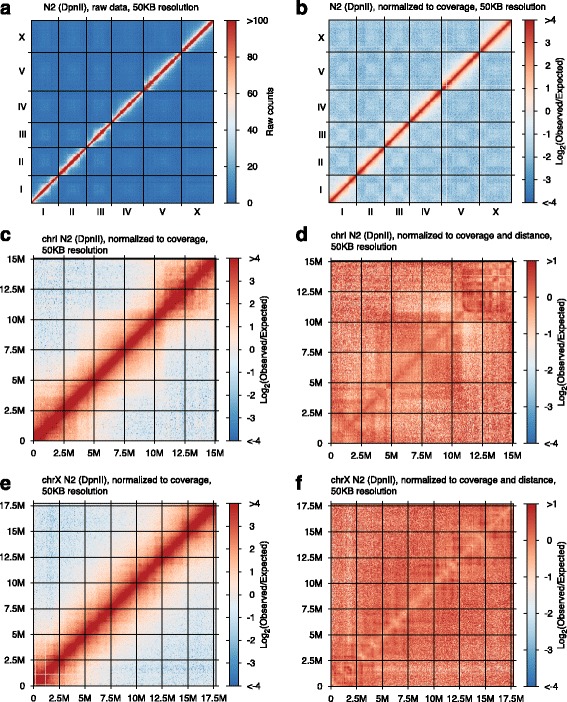
Results: Here we report a modified tethered conformation capture protocol that utilizes a series of rapid and efficient molecular manipulations. We applied the method to Caenorhabditis elegans, obtaining chromatin interaction maps that provide a sequence-anchored delineation of salient aspects of Caenorhabditis elegans chromosome structure, demonstrating a high level of consistency in overall chromosome organization between biological samples collected under different conditions. In addition to the application of the method to defining nuclear architecture, we found the resulting chromatin interaction maps to be of sufficient resolution and sensitivity to enable detection of large-scale structural variants such as inversions or translocations.
Conclusion: Our streamlined protocol provides an accelerated, robust, and broadly applicable means of generating chromatin spatial organization maps and detecting genome rearrangements without a need for cellular or chromatin fractionation.
Keywords: Caenorhabditis elegans; Chromatin; Conformation; Genome; Hi-C; TCC.
Figures




References
-
- Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, Sandstrom R, Bernstein B, Bender MA, Groudine M, Gnirke A, Stamatoyannopoulos J, Mirny LA, Lander ES, Dekker J. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009;326:289–293. doi: 10.1126/science.1181369. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
