

Using biological networks to integrate, visualize and analyze genomics data
- PMID: 27036106
- PMCID: PMC4818439
- DOI: 10.1186/s12711-016-0205-1
Using biological networks to integrate, visualize and analyze genomics data
Abstract
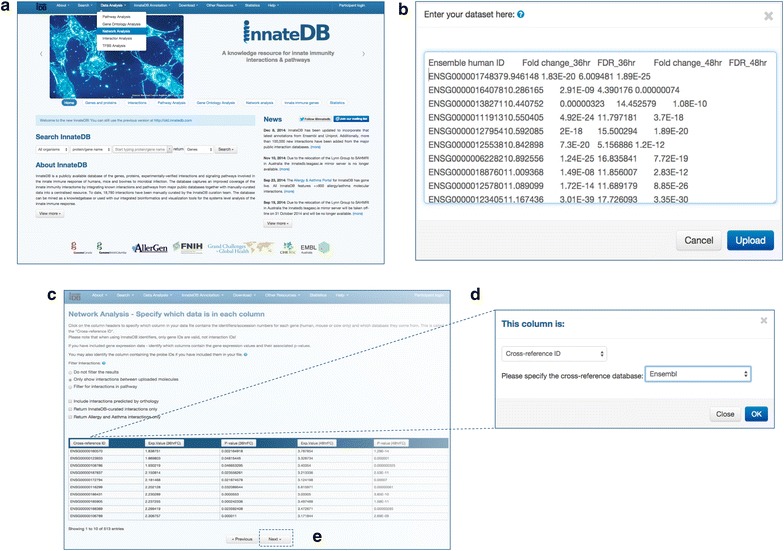
Network biology is a rapidly developing area of biomedical research and reflects the current view that complex phenotypes, such as disease susceptibility, are not the result of single gene mutations that act in isolation but are rather due to the perturbation of a gene's network context. Understanding the topology of these molecular interaction networks and identifying the molecules that play central roles in their structure and regulation is a key to understanding complex systems. The falling cost of next-generation sequencing is now enabling researchers to routinely catalogue the molecular components of these networks at a genome-wide scale and over a large number of different conditions. In this review, we describe how to use publicly available bioinformatics tools to integrate genome-wide 'omics' data into a network of experimentally-supported molecular interactions. In addition, we describe how to visualize and analyze these networks to identify topological features of likely functional relevance, including network hubs, bottlenecks and modules. We show that network biology provides a powerful conceptual approach to integrate and find patterns in genome-wide genomic data but we also discuss the limitations and caveats of these methods, of which researchers adopting these methods must remain aware.
Figures



References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
