

Hepatocyte growth factor as a downstream mediator of vascular endothelial growth factor-dependent preservation of growth in the developing lung
- PMID: 27036872
- PMCID: PMC4935471
- DOI: 10.1152/ajplung.00423.2015
Hepatocyte growth factor as a downstream mediator of vascular endothelial growth factor-dependent preservation of growth in the developing lung
Abstract
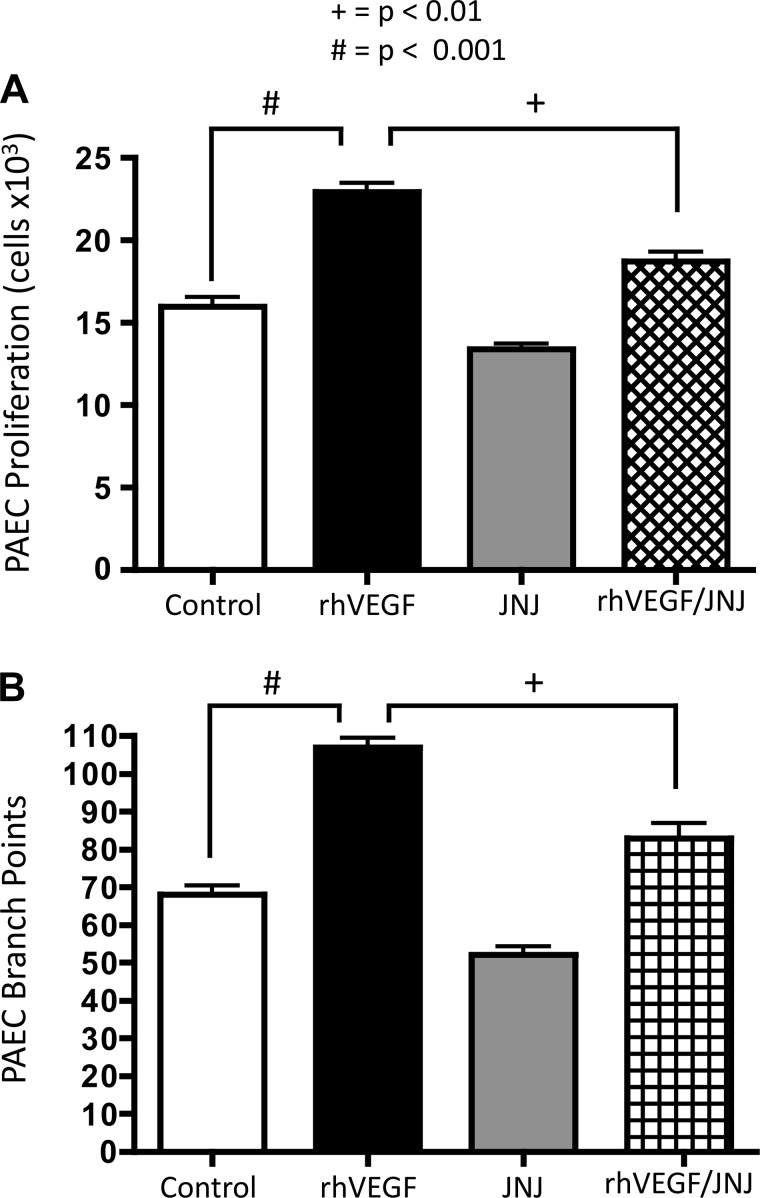
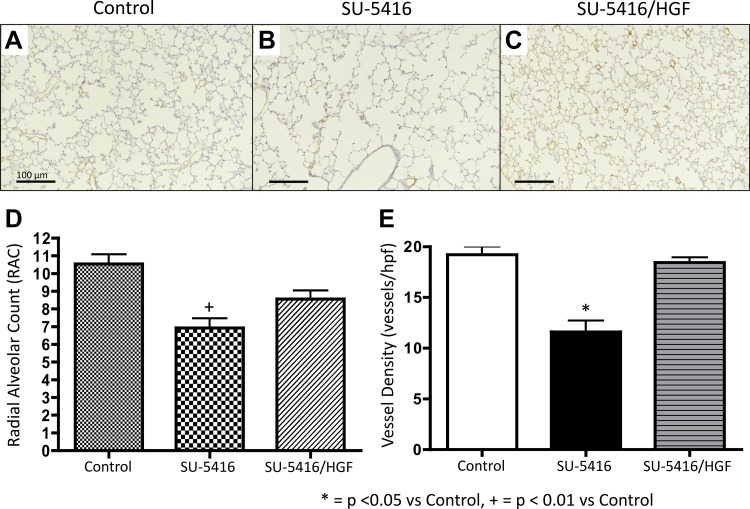
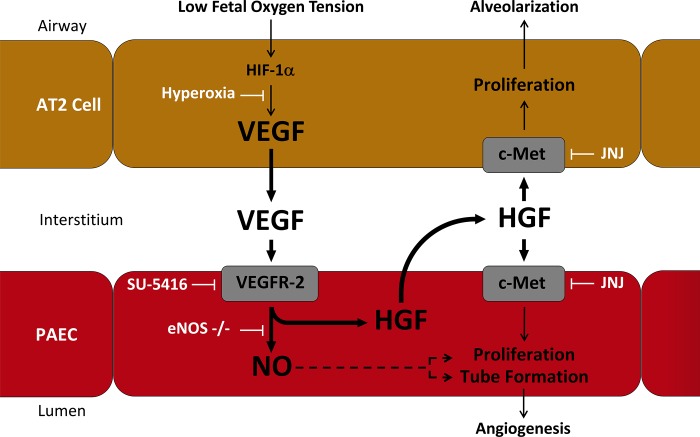
Impaired vascular endothelial growth factor (VEGF) signaling contributes to the pathogenesis of bronchopulmonary dysplasia (BPD). We hypothesized that the effects of VEGF on lung structure during development may be mediated through its downstream effects on both endothelial nitric oxide synthase (eNOS) and hepatocyte growth factor (HGF) activity, and that, in the absence of eNOS, trophic effects of VEGF would be mediated through HGF signaling. To test this hypothesis, we performed an integrative series of in vitro (fetal rat lung explants and isolated fetal alveolar and endothelial cells) and in vivo studies with normal rat pups and eNOS(-/-) mice. Compared with controls, fetal lung explants from eNOS(-/-) mice had decreased terminal lung bud formation, which was restored with recombinant human VEGF (rhVEGF) treatment. Neonatal eNOS(-/-) mice were more susceptible to hyperoxia-induced inhibition of lung growth than controls, which was prevented with rhVEGF treatment. Fetal alveolar type II (AT2) cell proliferation was increased with rhVEGF treatment only with mesenchymal cell (MC) coculture, and these effects were attenuated with anti-HGF antibody treatment. Unlike VEGF, HGF directly stimulated isolated AT2 cells even without MC coculture. HGF directly stimulates fetal pulmonary artery endothelial cell growth and tube formation, which is attenuated by treatment with JNJ-38877605, a c-Met inhibitor. rHGF treatment preserves alveolar and vascular growth after postnatal exposure to SU-5416, a VEGF receptor inhibitor. We conclude that the effects of VEGF on AT2 and endothelial cells during lung development are partly mediated through HGF-c-Met signaling and speculate that reciprocal VEGF-HGF signaling between epithelia and endothelia is disrupted in infants who develop BPD.
Keywords: bronchopulmonary dysplasia; endothelial nitric oxide synthase; hepatocyte growth factor; lung development; lung hypoplasia; nitric oxide; vascular endothelial growth factor.
Copyright © 2016 the American Physiological Society.
Figures








Similar articles
-
Recombinant human VEGF treatment transiently increases lung edema but enhances lung structure after neonatal hyperoxia.Am J Physiol Lung Cell Mol Physiol. 2006 Nov;291(5):L1068-78. doi: 10.1152/ajplung.00093.2006. Epub 2006 Jul 7. Am J Physiol Lung Cell Mol Physiol. 2006. PMID: 16829629
-
[Dynamic changes in vascular endothelial growth factor and endothelial nitric oxide synthase in lungs of premature rats after hyperoxia exposure].Zhongguo Dang Dai Er Ke Za Zhi. 2007 Oct;9(5):473-8. Zhongguo Dang Dai Er Ke Za Zhi. 2007. PMID: 17937862 Chinese.
-
Inhaled nitric oxide attenuates pulmonary hypertension and improves lung growth in infant rats after neonatal treatment with a VEGF receptor inhibitor.Am J Physiol Lung Cell Mol Physiol. 2004 Aug;287(2):L344-51. doi: 10.1152/ajplung.00291.2003. Epub 2004 Apr 2. Am J Physiol Lung Cell Mol Physiol. 2004. PMID: 15064225
-
Lung epithelial-endothelial-mesenchymal signaling network with hepatocyte growth factor as a hub is involved in bronchopulmonary dysplasia.Front Cell Dev Biol. 2024 Sep 3;12:1462841. doi: 10.3389/fcell.2024.1462841. eCollection 2024. Front Cell Dev Biol. 2024. PMID: 39291265 Free PMC article. Review.
-
Vascular endothelial growth factor of the lung: friend or foe.Curr Opin Pharmacol. 2008 Jun;8(3):255-60. doi: 10.1016/j.coph.2008.03.003. Epub 2008 May 28. Curr Opin Pharmacol. 2008. PMID: 18468486 Free PMC article. Review.
Cited by
-
Unique aspects of the developing lung circulation: structural development and regulation of vasomotor tone.Pulm Circ. 2016 Dec;6(4):407-425. doi: 10.1086/688890. Pulm Circ. 2016. PMID: 27942377 Free PMC article. Review.
-
Targeting the lung endothelial niche to promote angiogenesis and regeneration: A review of applications.Front Mol Biosci. 2022 Dec 19;9:1093369. doi: 10.3389/fmolb.2022.1093369. eCollection 2022. Front Mol Biosci. 2022. PMID: 36601582 Free PMC article. Review.
-
Investigation of the miRNA-mRNA Regulatory Circuits and Immune Signatures Associated with Bronchopulmonary Dysplasia.J Inflamm Res. 2024 Mar 5;17:1467-1480. doi: 10.2147/JIR.S448394. eCollection 2024. J Inflamm Res. 2024. PMID: 38476468 Free PMC article.
-
Hedgehog-responsive PDGFRa(+) fibroblasts maintain a unique pool of alveolar epithelial progenitor cells during alveologenesis.Cell Rep. 2022 Apr 5;39(1):110608. doi: 10.1016/j.celrep.2022.110608. Cell Rep. 2022. PMID: 35385750 Free PMC article.
-
Mesodermal lineages in the developing respiratory system.Trends Dev Biol. 2016;9:91-110. Trends Dev Biol. 2016. PMID: 34707332 Free PMC article.
References
-
- Abman SH. Bronchopulmonary Dysplasia. New York: Informa Healthcare, 2010, p. xvii.
-
- Abman SH. Impaired vascular endothelial growth factor signaling in the pathogenesis of neonatal pulmonary vascular disease. In: Membrane Receptors, Channels and Transporters in Pulmonary Circulation, edited by Yuan JXJ and Ward JPT. New York: Humana, 2010, p. 323–335. - PubMed
-
- Balasubramaniam V. Nitric oxide augments fetal pulmonary artery endothelial cell angiogenesis in vitro. Am J Physiol Lung Cell Mol Physiol 290: L1111–L1116, 2006. - PubMed
-
- Balasubramaniam V, Maxey A, Abman S. Inhaled nitric oxide reverses hypoxia induced lung hypoplasia in endothelial nitric oxide synthase-deficient mice. Chest 128: 613S–614S, 2005. - PubMed
-
- Balasubramaniam V, Maxey AM, Morgan DB, Markham NE, Abman SH. Inhaled NO restores lung structure in eNOS-deficient mice recovering from neonatal hypoxia. Am J Physiol Lung Cell Mol Physiol 291: L119–L127, 2006. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous