

Review
doi: 10.1101/cshperspect.a019521.
Histone Modifications and Cancer
Affiliations
- PMID: 27037415
- PMCID: PMC4817802
- DOI: 10.1101/cshperspect.a019521
Item in Clipboard
Review
Histone Modifications and Cancer
Cold Spring Harb Perspect Biol.
.
Abstract
Histone posttranslational modifications represent a versatile set of epigenetic marks involved not only in dynamic cellular processes, such as transcription and DNA repair, but also in the stable maintenance of repressive chromatin. In this article, we review many of the key and newly identified histone modifications known to be deregulated in cancer and how this impacts function. The latter part of the article addresses the challenges and current status of the epigenetic drug development process as it applies to cancer therapeutics.
Copyright © 2016 Cold Spring Harbor Laboratory Press; all rights reserved.
Figures
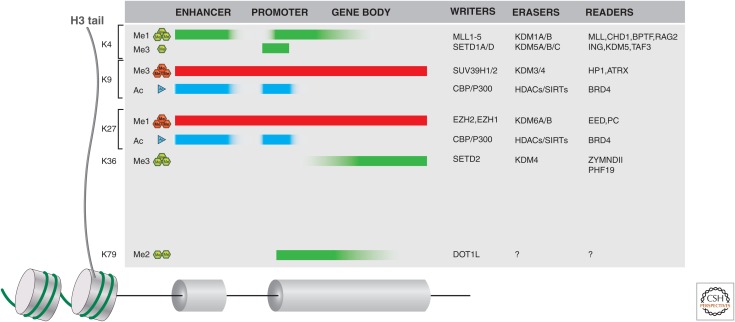
Histone writers, erasers, and readers in cancer. Histone H3 tail lysine residues, frequently subject to posttranslational modifications (PTMs), are indicated along the left side. The typical distribution of these H3 PTMs is also indicated along the length of gene loci (including distal enhancers) as shaded blocks. Green (methylation) or cyan (acetylation) indicates histone marks associated with active genes, whereas red shading is indicative of silent genes. A few examples of writers, erasers, and readers that may propagate a mark or act as an effector protein are listed on the right side of the figure. For a more complete listing of these proteins, see Appendices A–D at the end of this article.
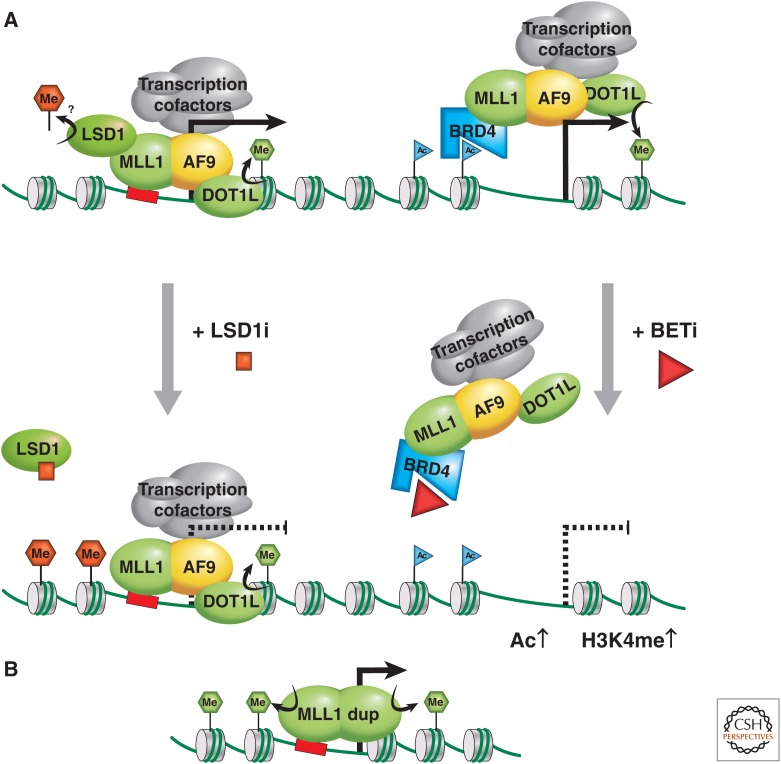
MLL as an oncogene. (A) The MLL1-AF9 (or ENL) fusion oncoprotein activates transcription via two possible mechanisms; the left mechanism attributes recruitment of MLL1-AF9 to chromatin via the MLL1 portion, and transcription is activated via association with cofactors, including the DOT1 methyltransferase, methylating H3K79 (green hexagon) and the pTEFb complex, which modifies RNA Polymerase II into the active elongating form. (B) The partial duplication of an MLL gene can result in duplication of an internal region that includes chromatin binding features and protein–protein interaction domains, providing oncogenic methyltransferase H3K4me3 activity and increased transcriptional activation. LSD1 may be involved via the MLL supercomplex, or a transcription elongation complex contributing to oncogenic activity through H4K4me2 or H3K9me2 demethylase activity. LSD1 inhibition somehow reduces the oncogenic program, promoting differentiation. In the mechanism on the right, recruitment of MLL1-AF9 to chromatin is attributed to its association with BRD-2, -3, or -4 via acetylated chromatin. This mechanism of gene activation can therapeutically be targeted via treatment with BET inhibitors.
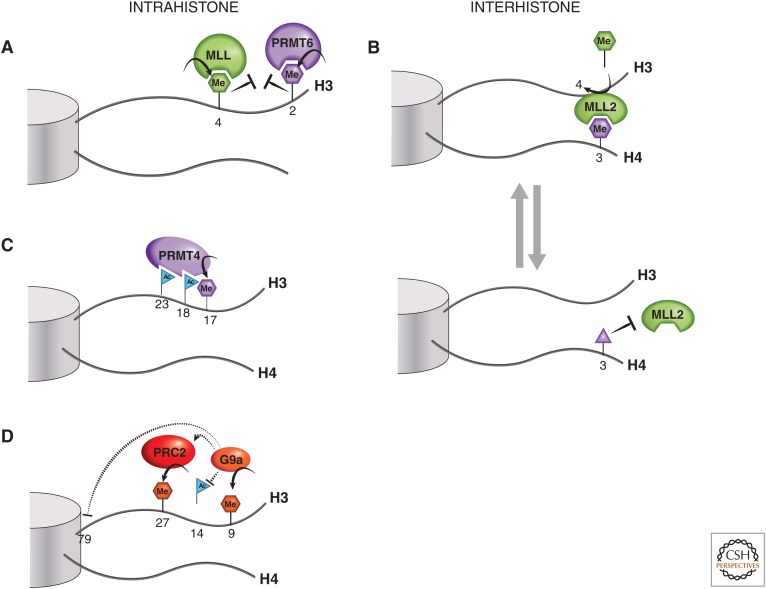
Histone tail cross talk. (A) H3R2me2a and H3K4 methylation are examples of mutually exclusive intrahistone H3 PTMs. (B) An example of an interhistone cross talk is when H4R3me0 or H4R3me2a marks (purple hexagon) are converted into H4R3me2s (purple triangle), which is thought to block the binding of MLL2 via its PHD4-6 domain, thus preventing the methyltransferase activity of MLL2 at H3K4. (C) PRMT4, a histone arginine methyltransferase, is thought to partially rely on H3 acetylation at K18 and K23 for recruitment to H3 and subsequent dimethylation of the nearby R17 residue. (D) An illustration of the increasingly complex picture of histone H3 tail cross talk, involving H3K9, H3K27, and H3K79 methylation and H3K14 acetylation. See text for a more detailed explanation.
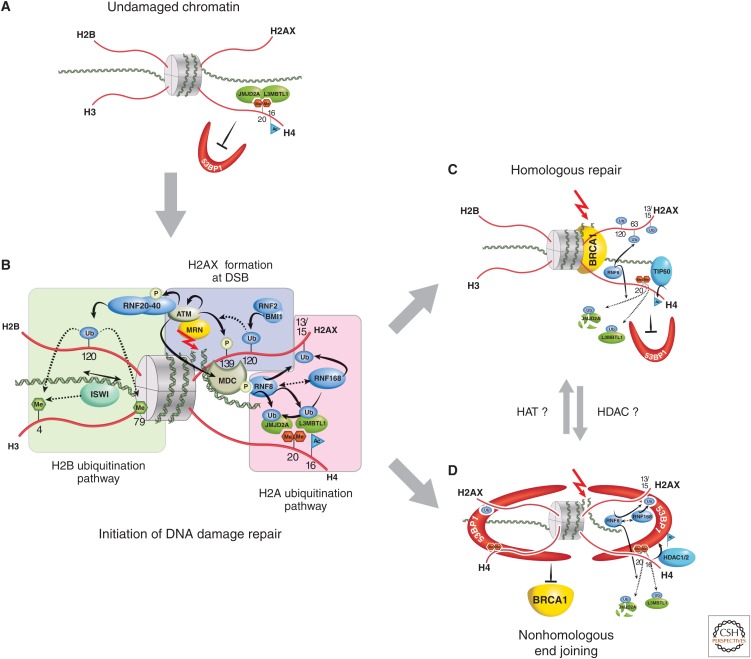
Role of histone PTMs in DNA damage repair. (A) Before DNA damage, L3MBTL1 and JMJD2A (possibly also JMJD2B) bind, via their Tudor domains, to H4K20me2 (red hexagons), hindering access by DNA repair proteins, such as 53BP1. (B) When double-strand DNA breaks occur (red arrow), the DNA damage–sensing proteins, such as MRN, act to initiate cascades of protein and histone tail PTMs (blue-shaded area). In particular, the phosphorylation of H2A at S139 represents the conversion of H2A into H2A.X. Subsequent chromatin-remodeling events occur as a consequence of H2B ubiquitination (green-shaded area), whereas further ubiquitination events on H2A.X alter local chromatin structure (pink-shaded area). This latter RNF8/RNF168-catalyzed H2A ubiquitination pathway also polyubiquitinates JMJD2A/B for degradation, whereas L3MBTL1 is removed by RNF8-mediated ubiquitination. This then allows DSBs in DNA to be repaired by homologous recombinant repair (HRR), where joining occurs between two similar or identical strands of DNA (C); or by nonhomologous end joining (NHEJ), in which the two DNA ends are joined directly, usually with no sequence homology, although, in some cases, regions of microhomology are used (D). (C) During HRR, TIP60 acetylates H4K16, RNF8 ubiquitinates H2AK63, and selectively allows for BRCA1 binding but not 53BP1. (D) If NHEJ is required, H4K16 is deacetylated (presumably by HDAC1,2) and H2AK15 is ubiquitinated. 53BP1 binds to both H4K20me2 and H2AK15ub, whereas BRCA1 is excluded. To illustrate 53BP1 oligmer formation, only histone H4 and H2AX tails are illustrated in this panel.
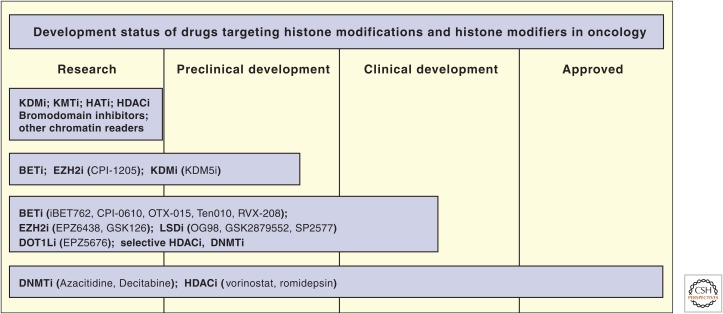
The development status of epigenetic drugs. KDMi, lysine demethylase inhibitor; KMTi, lysine methyltransferase inhibitor; HATi, histone acetyltransferase inhibitor; HDACi, histone deacetylase inhibitor; BETi, bromodomain and extracarboxy terminal domain inhibitor; DNMTi, DNA methyltransferase inhibitor.
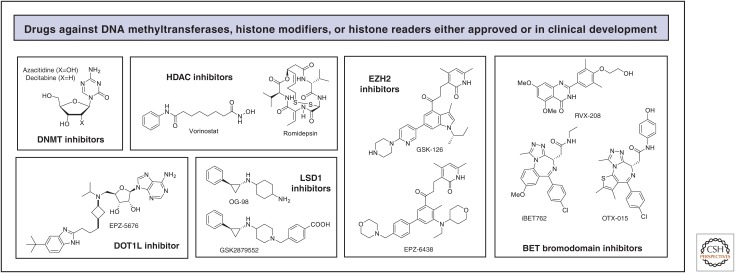
Representative structures of drugs targeting DNA methylation, histone modifications, and histone readers in oncology.
References
-
- Allis CD, Jenuwein T, Reinberg D. 2014. Overview and concepts. Cold Spring Harb Perspect Biol 10.1101/cshperspect.a018739. - DOI
-
- Anderton JA, Bose S, Vockerodt M, Vrzalikova K, Wei W, Kuo M, Helin K, Christensen J, Rowe M, Murray PG, et al. 2011. The H3K27me3 demethylase, KDM6B, is induced by Epstein-Barr virus and over-expressed in Hodgkin’s Lymphoma. Oncogene 30: 2037–2043. - PubMed
-
- Angrand PO, Apiou F, Stewart AF, Dutrillaux B, Losson R, Chambon P. 2001. NSD3, a new SET domain-containing gene, maps to 8p12 and is amplified in human breast cancer cell lines. Genomics 74: 79–88. - PubMed
-
- Arrowsmith CH, Bountra C, Fish PV, Lee K, Schapira M. 2012. Epigenetic protein families: A new frontier for drug discovery. Nat Rev Drug Discov 11: 384–400. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources