

rAAV Gene Therapy in a Canavan's Disease Mouse Model Reveals Immune Impairments and an Extended Pathology Beyond the Central Nervous System
- PMID: 27039844
- PMCID: PMC4923332
- DOI: 10.1038/mt.2016.68
rAAV Gene Therapy in a Canavan's Disease Mouse Model Reveals Immune Impairments and an Extended Pathology Beyond the Central Nervous System
Abstract
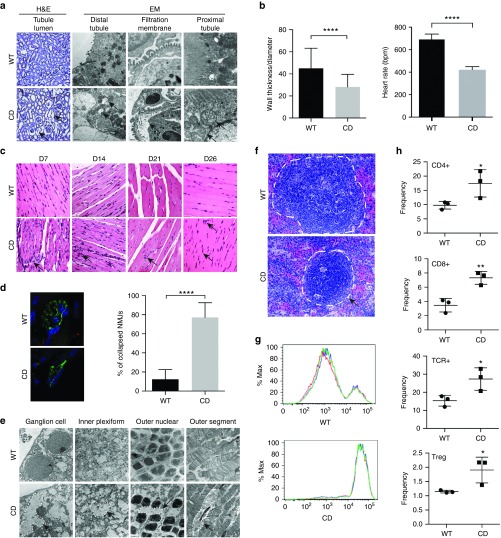
Aspartoacylase (AspA) gene mutations cause the pediatric lethal neurodegenerative Canavan disease (CD). There is emerging promise of successful gene therapy for CD using recombinant adeno-associated viruses (rAAVs). Here, we report an intracerebroventricularly delivered AspA gene therapy regime using three serotypes of rAAVs at a 20-fold reduced dose than previously described in AspA(-/-) mice, a bona-fide mouse model of CD. Interestingly, central nervous system (CNS)-restricted therapy prolonged survival over systemic therapy in CD mice but failed to sustain motor functions seen in systemically treated mice. Importantly, we reveal through histological and functional examination of untreated CD mice that AspA deficiency in peripheral tissues causes morphological and functional abnormalities in this heretofore CNS-defined disorder. We demonstrate for the first time that AspA deficiency, possibly through excessive N-acetyl aspartic acid accumulation, elicits both a peripheral and CNS immune response in CD mice. Our data establish a role for peripheral tissues in CD pathology and serve to aid the development of more efficacious and sustained gene therapy for this disease.
Figures




References
-
- Traeger, EC and Rapin, I (1998). The clinical course of Canavan disease. Pediatr Neurol 18: 207–212. - PubMed
-
- Kaul, R, Gao, GP, Balamurugan, K and Matalon, R (1993). Cloning of the human aspartoacylase cDNA and a common missense mutation in Canavan disease. Nat Genet 5: 118–123. - PubMed
-
- Matalon, R, Michals, K, Sebesta, D, Deanching, M, Gashkoff, P and Casanova, J (1988). Aspartoacylase deficiency and N-acetylaspartic aciduria in patients with Canavan disease. Am J Med Genet 29: 463–471. - PubMed
-
- Birnbaum, SM, Levintow, L, Kingsley, RB and Greenstein, JP (1952). Specificity of amino acid acylases. J Biol Chem 194: 455–470. - PubMed
-
- Mehta, V and Namboodiri, MA (1995). N-acetylaspartate as an acetyl source in the nervous system. Brain Res Mol Brain Res 31: 151–157. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
