

Recessive Inactivating Mutations in TBCK, Encoding a Rab GTPase-Activating Protein, Cause Severe Infantile Syndromic Encephalopathy
- PMID: 27040692
- PMCID: PMC4833196
- DOI: 10.1016/j.ajhg.2016.01.016
Recessive Inactivating Mutations in TBCK, Encoding a Rab GTPase-Activating Protein, Cause Severe Infantile Syndromic Encephalopathy
Abstract
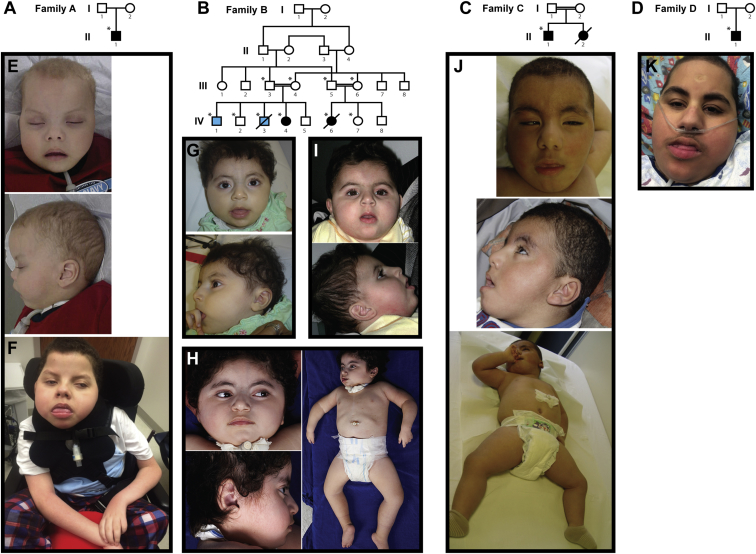
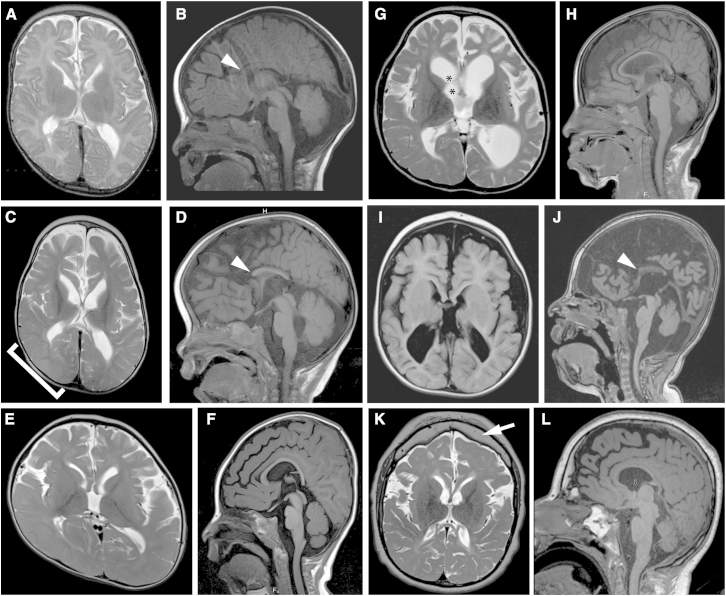
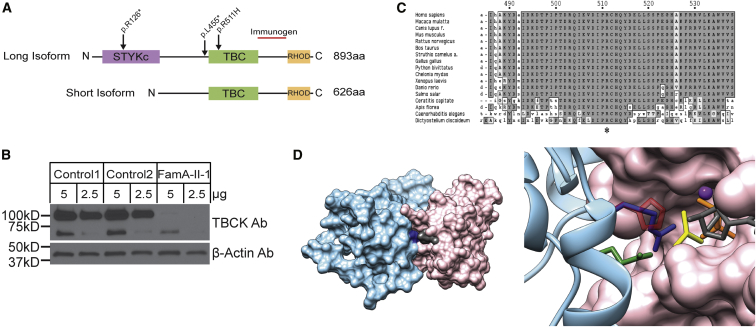
Infantile encephalopathies are a group of clinically and biologically heterogeneous disorders for which the genetic basis remains largely unknown. Here, we report a syndromic neonatal encephalopathy characterized by profound developmental disability, severe hypotonia, seizures, diminished respiratory drive requiring mechanical ventilation, brain atrophy, dysgenesis of the corpus callosum, cerebellar vermis hypoplasia, and facial dysmorphism. Biallelic inactivating mutations in TBCK (TBC1-domain-containing kinase) were independently identified by whole-exome sequencing as the cause of this condition in four unrelated families. Matching these families was facilitated by the sharing of phenotypic profiles and WES data in a recently released web-based tool (Geno2MP) that links phenotypic information to rare variants in families with Mendelian traits. TBCK is a putative GTPase-activating protein (GAP) for small GTPases of the Rab family and has been shown to control cell growth and proliferation, actin-cytoskeleton dynamics, and mTOR signaling. Two of the three mutations (c.376C>T [p.Arg126(∗)] and c.1363A>T [p.Lys455(∗)]) are predicted to truncate the protein, and loss of the major TBCK isoform was confirmed in primary fibroblasts from one affected individual. The third mutation, c.1532G>A (p.Arg511His), alters a conserved residue within the TBC1 domain. Structural analysis implicated Arg511 as a required residue for Rab-GAP function, and in silico homology modeling predicted impaired GAP function in the corresponding mutant. These results suggest that loss of Rab-GAP activity is the underlying mechanism of disease. In contrast to other disorders caused by dysregulated mTOR signaling associated with focal or global brain overgrowth, impaired TBCK function results in progressive loss of brain volume.
Keywords: GTPase-activating protein; Mendelian disease; exome sequencing; infantile encephalopathy; mTOR signaling.
Copyright © 2016 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Hunt D., Leventer R.J., Simons C., Taft R., Swoboda K.J., Gawne-Cain M., Magee A.C., Turnpenny P.D., Baralle D., DDD study Whole exome sequencing in family trios reveals de novo mutations in PURA as a cause of severe neurodevelopmental delay and learning disability. J. Med. Genet. 2014;51:806–813. - PMC - PubMed
-
- Perez Y., Kadir R., Volodarsky M., Noyman I., Flusser H., Shorer Z., Gradstein L., Birnbaum R.Y., Birk O.S. UNC80 mutation causes a syndrome of hypotonia, severe intellectual disability, dyskinesia and dysmorphism, similar to that caused by mutations in its interacting cation channel NALCN. J. Med. Genet. 2015 - PubMed
-
- Stray-Pedersen A., Cobben J.-M., Prescott T.E., Lee S., Cang C., Aranda K., Ahmed S., Alders M., Gerstner T., Aslaksen K., Care4Rare Canada Consortium. Baylor-Hopkins Center for Mendelian Genomics Biallelic Mutations in UNC80 Cause Persistent Hypotonia, Encephalopathy, Growth Retardation, and Severe Intellectual Disability. Am. J. Hum. Genet. 2016;98:202–209. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
