

Ambroxol chaperone therapy for neuronopathic Gaucher disease: A pilot study
- PMID: 27042680
- PMCID: PMC4774255
- DOI: 10.1002/acn3.292
Ambroxol chaperone therapy for neuronopathic Gaucher disease: A pilot study
Abstract
Objective: Gaucher disease (GD) is a lysosomal storage disease characterized by a deficiency of glucocerebrosidase. Although enzyme-replacement and substrate-reduction therapies are available, their efficacies in treating the neurological manifestations of GD are negligible. Pharmacological chaperone therapy is hypothesized to offer a new strategy for treating the neurological manifestations of this disease. Specifically, ambroxol, a commonly used expectorant, has been proposed as a candidate pharmacological chaperone. The purpose of this study was to evaluate the safety, tolerability, and neurological efficacy of ambroxol in patients with neuronopathic GD.
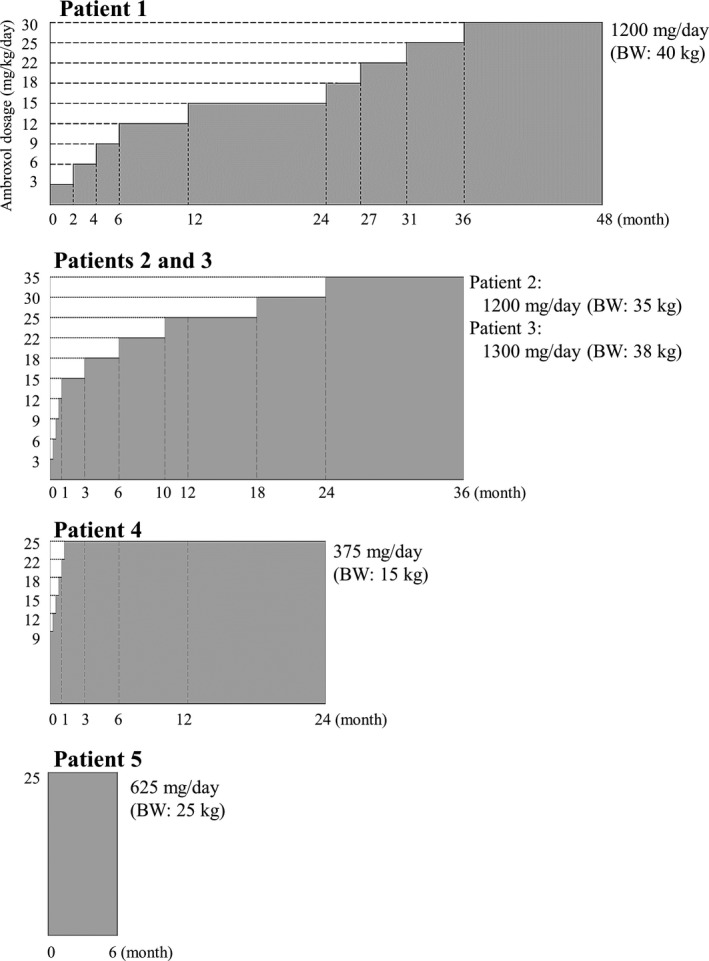
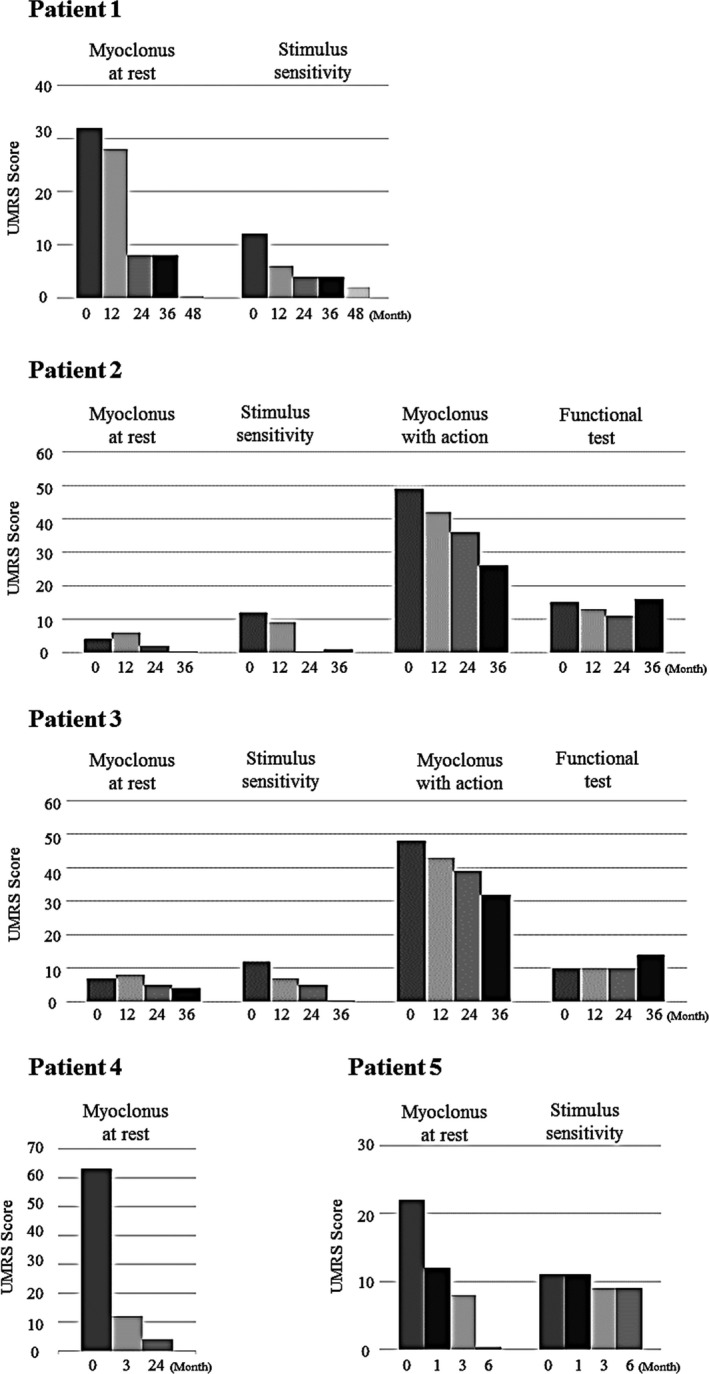
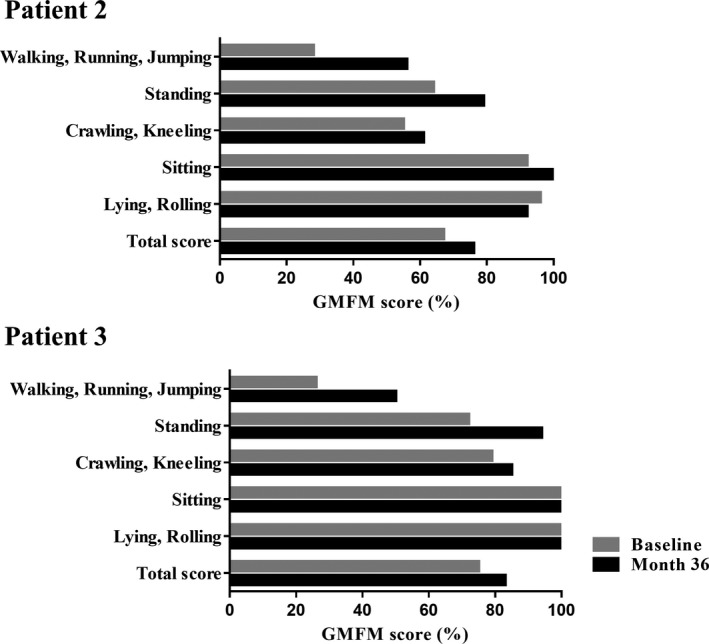
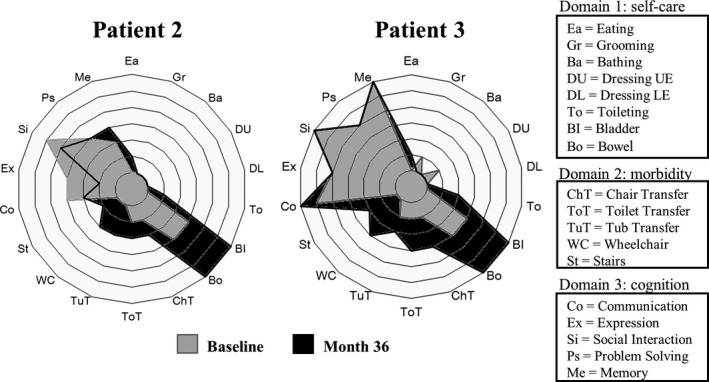
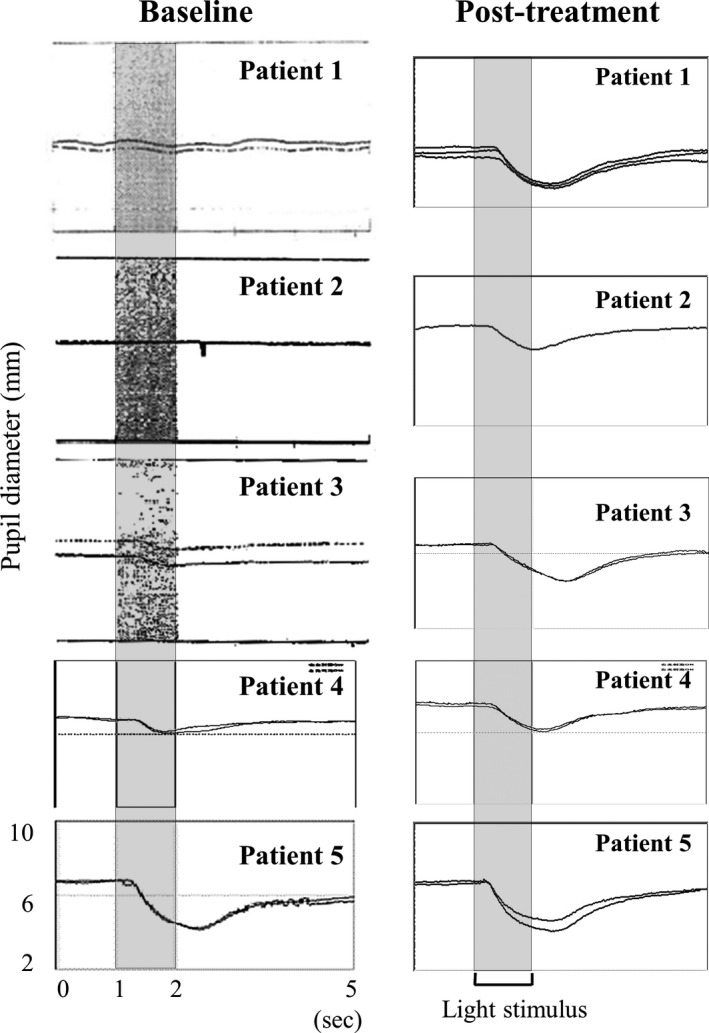
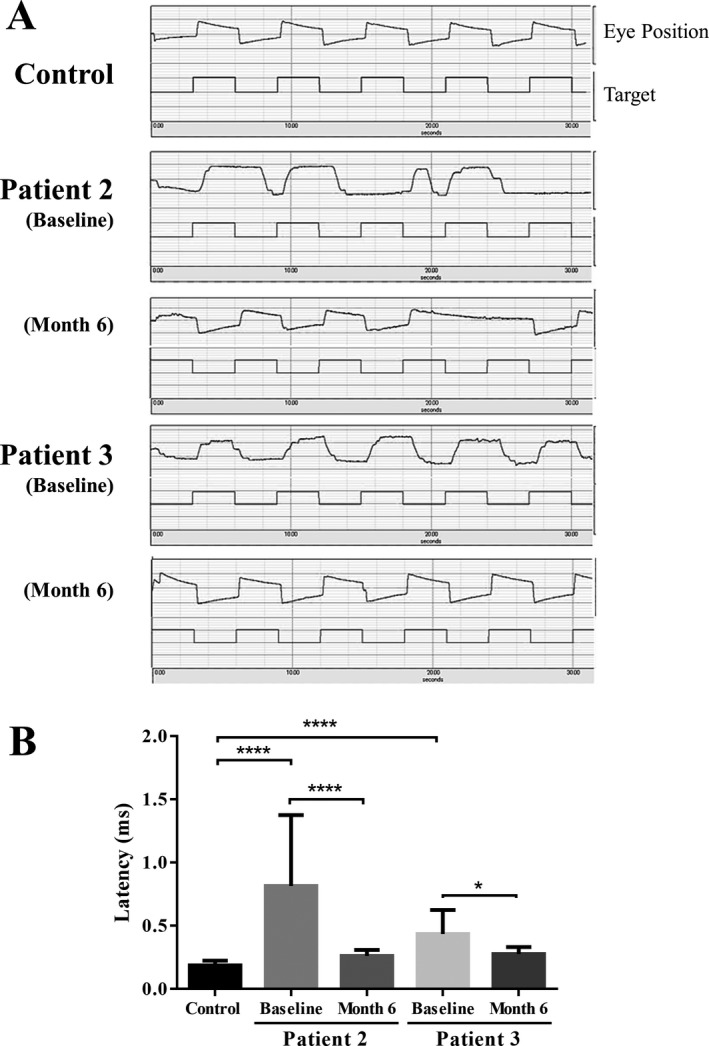
Methods: This open-label pilot study included five patients who received high-dose oral ambroxol in combination with enzyme replacement therapy. Safety was assessed by adverse event query, physical examination, electrocardiography, laboratory studies, and drug concentration. Biochemical efficacy was assessed through evidence of glucocerebrosidase activity in the lymphocytes and glucosylsphingosine levels in the cerebrospinal fluid. Neurological efficacy was evaluated using the Unified Myoclonus Rating Scale, Gross Motor Function Measure, Functional Independence Measure, seizure frequency, pupillary light reflex, horizontal saccadic latency, and electrophysiologic studies.
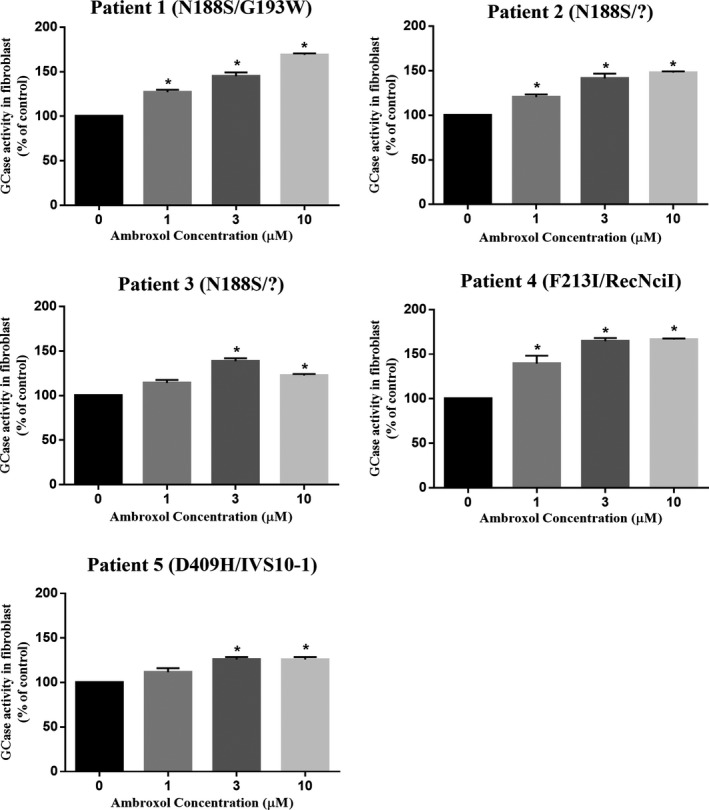
Results: High-dose oral ambroxol had good safety and tolerability, significantly increased lymphocyte glucocerebrosidase activity, permeated the blood-brain barrier, and decreased glucosylsphingosine levels in the cerebrospinal fluid. Myoclonus, seizures, and pupillary light reflex dysfunction markedly improved in all patients. Relief from myoclonus led to impressive recovery of gross motor function in two patients, allowing them to walk again.
Interpretation: Pharmacological chaperone therapy with high-dose oral ambroxol shows promise in treating neuronopathic GD, necessitating further clinical trials.
Figures
References
-
- Orvisky E, Park JK, LaMarca ME, et al. Glucosylsphingosine accumulation in tissues from patients with Gaucher disease: correlation with phenotype and genotype. Mol Genet Metab 2002;76:262–270. - PubMed
-
- Schueler UH, Kolter T, Kaneski CR, et al. Toxicity of glucosylsphingosine (glucopsychosine) to cultured neuronal cells: a model system for assessing neuronal damage in Gaucher disease type 2 and 3. Neurobiol Dis 2003;14:595–601. - PubMed
-
- Nilsson O, Svennerholm L. Accumulation of glucosylceramide and glucosylsphingosine (psychosine) in cerebrum and cerebellum in infantile and juvenile Gaucher disease. J Neurochem 1982;39:709–718. - PubMed
-
- Grabowski GA, Petsko GA, Kolodny EH. The Online Metabolic and Molecular Basis of Inherited Disease (OMMBID) Chapter 146, 2014.
-
- Grabowski GA. Phenotype, diagnosis, and treatment of Gaucher's disease. Lancet 2008;372:1263–12671. - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
