

Targeting mitochondrial biogenesis to overcome drug resistance to MAPK inhibitors
- PMID: 27043285
- PMCID: PMC4855947
- DOI: 10.1172/JCI82661
Targeting mitochondrial biogenesis to overcome drug resistance to MAPK inhibitors
Abstract
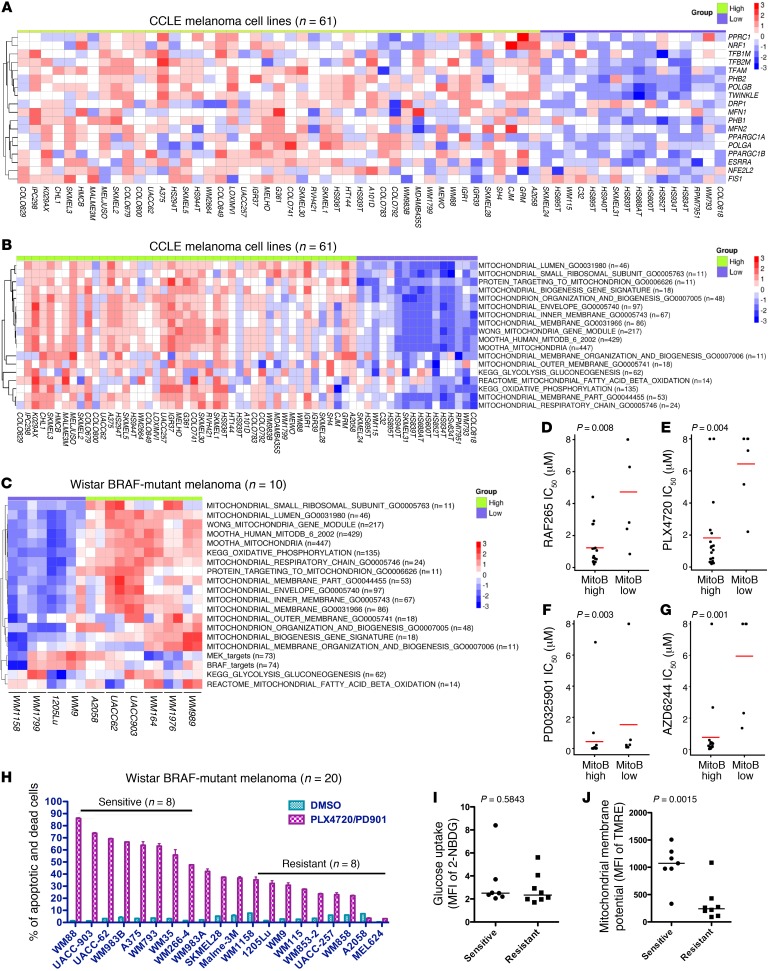
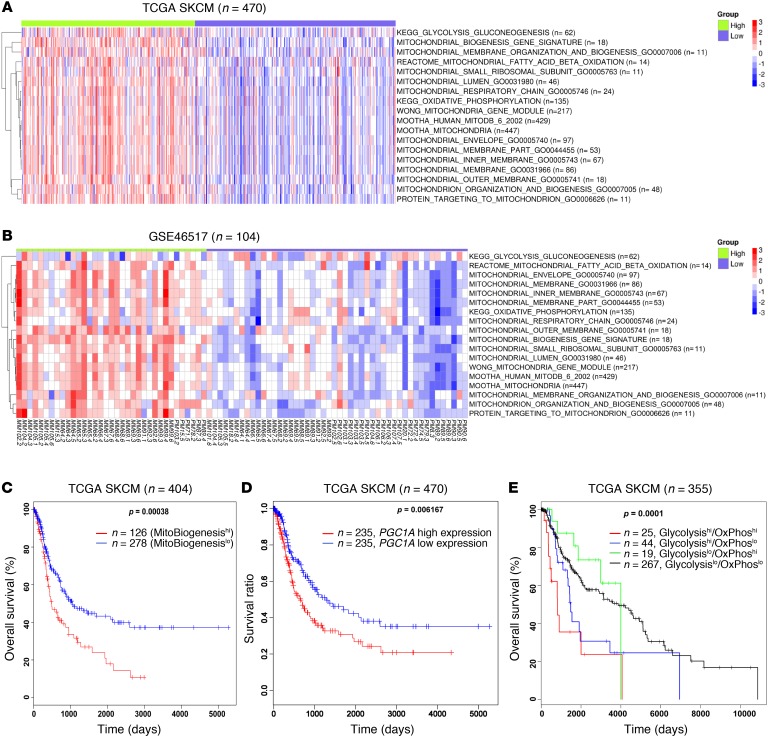
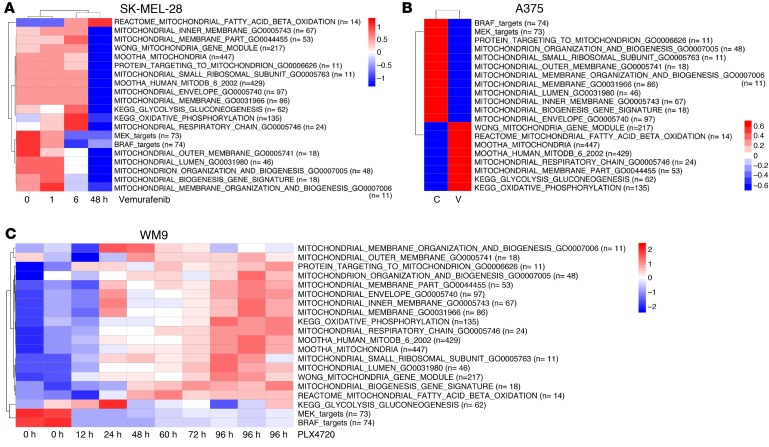
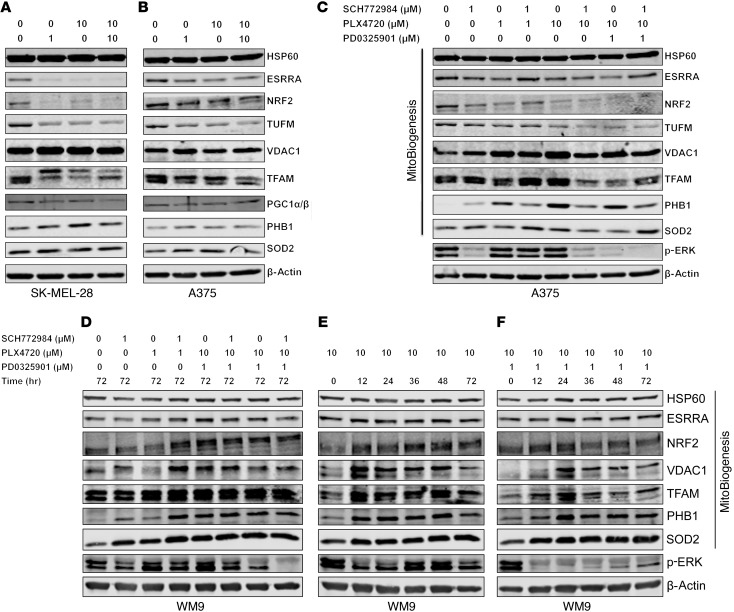
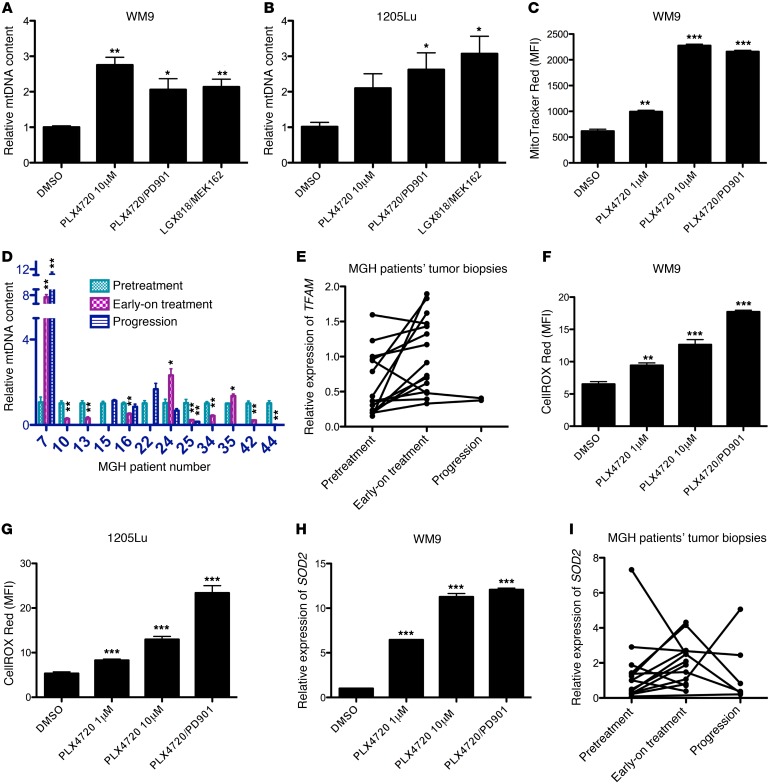
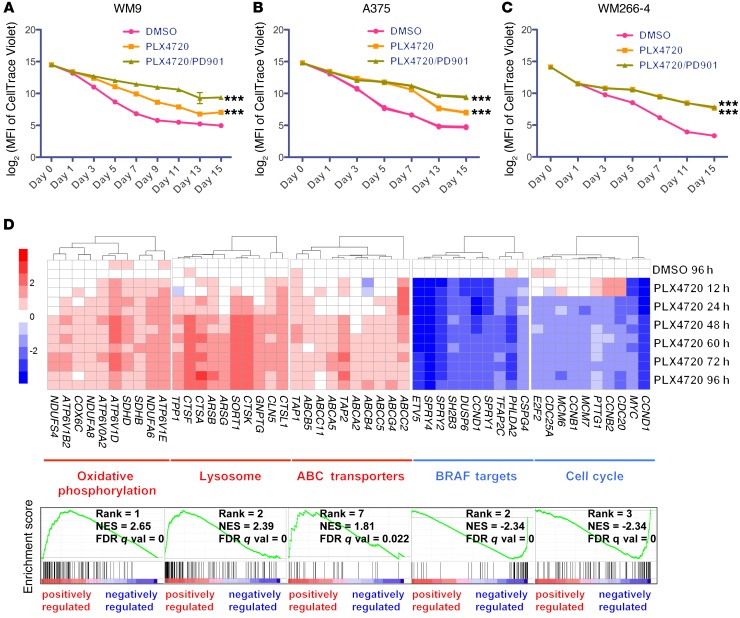
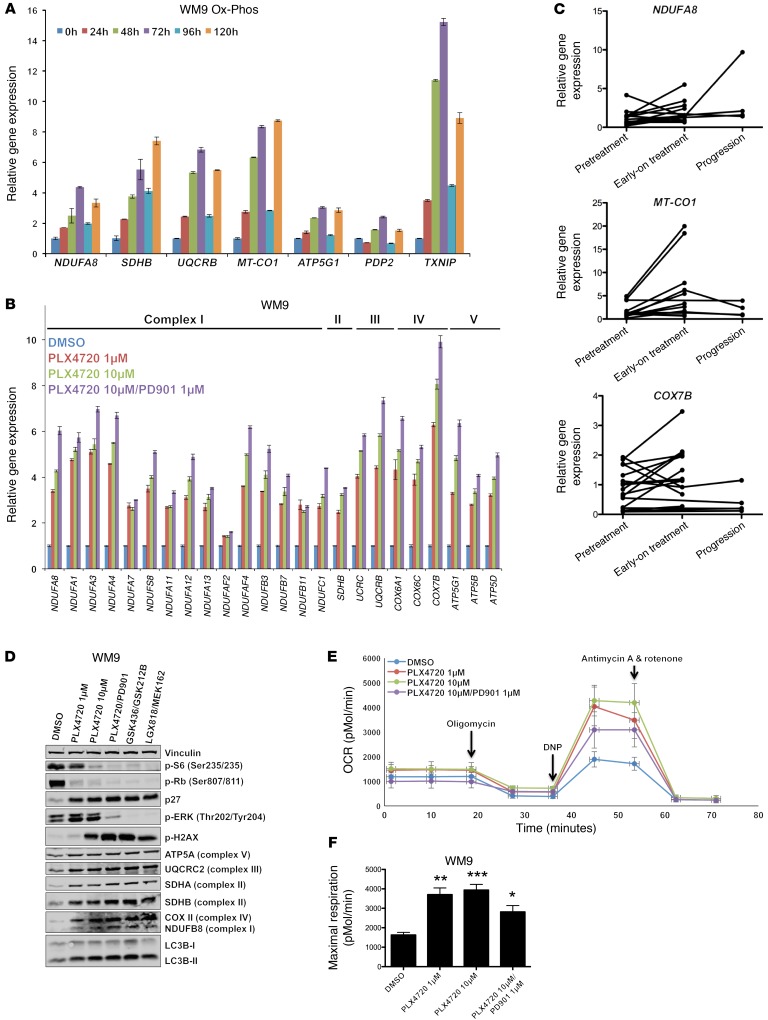
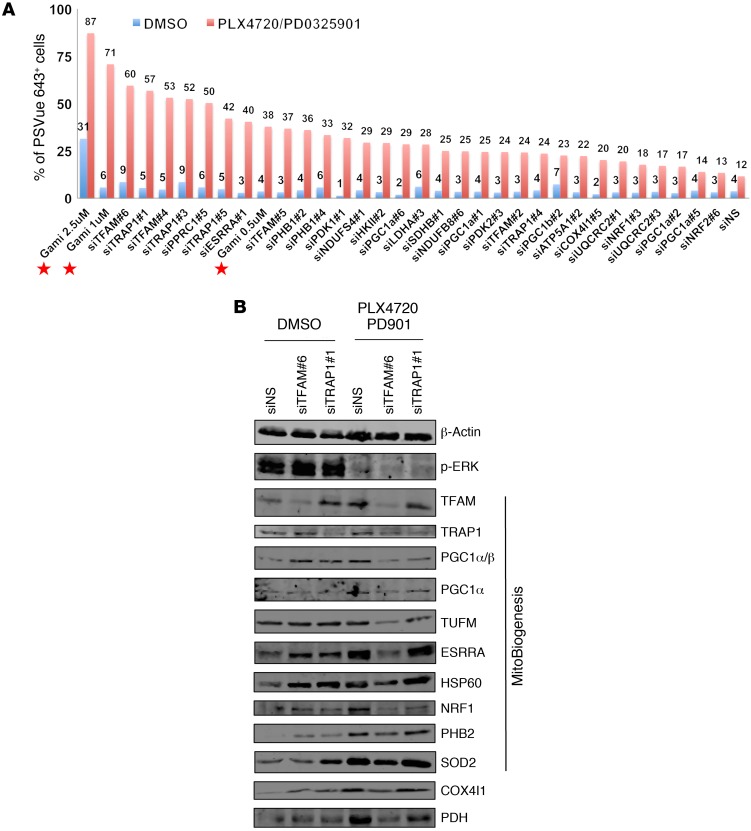
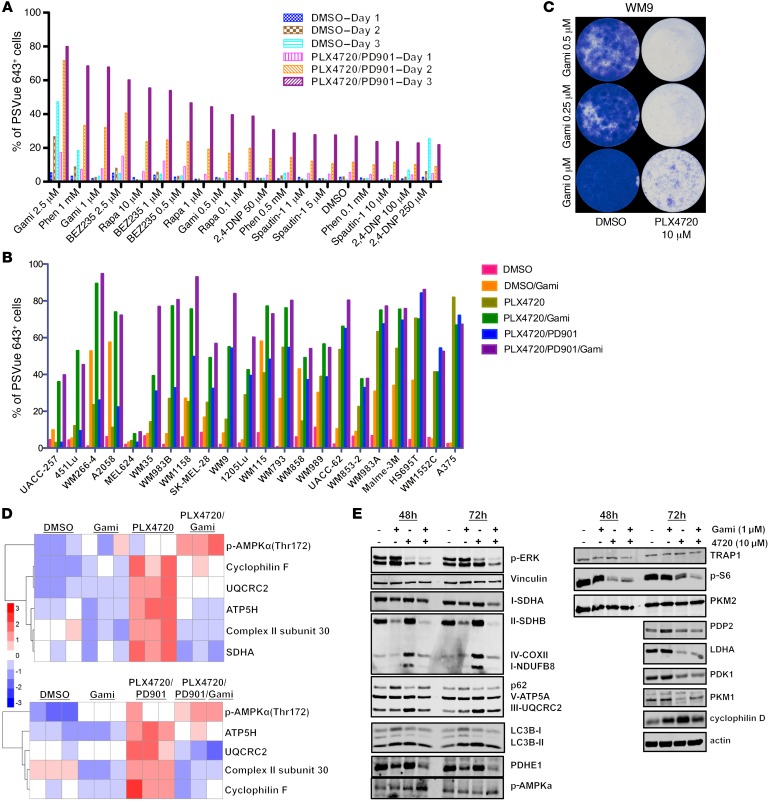
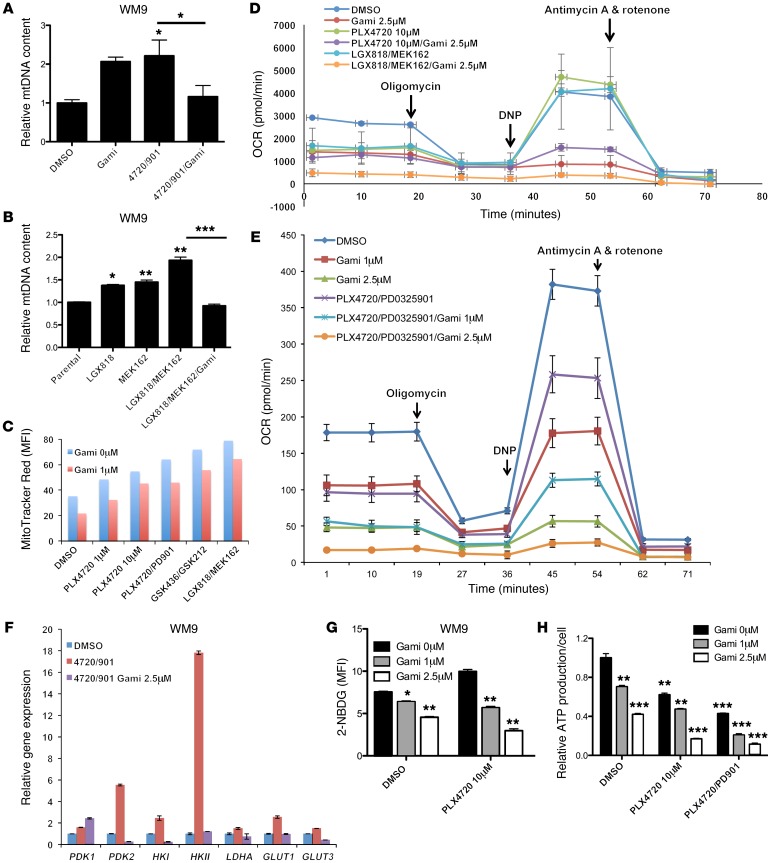
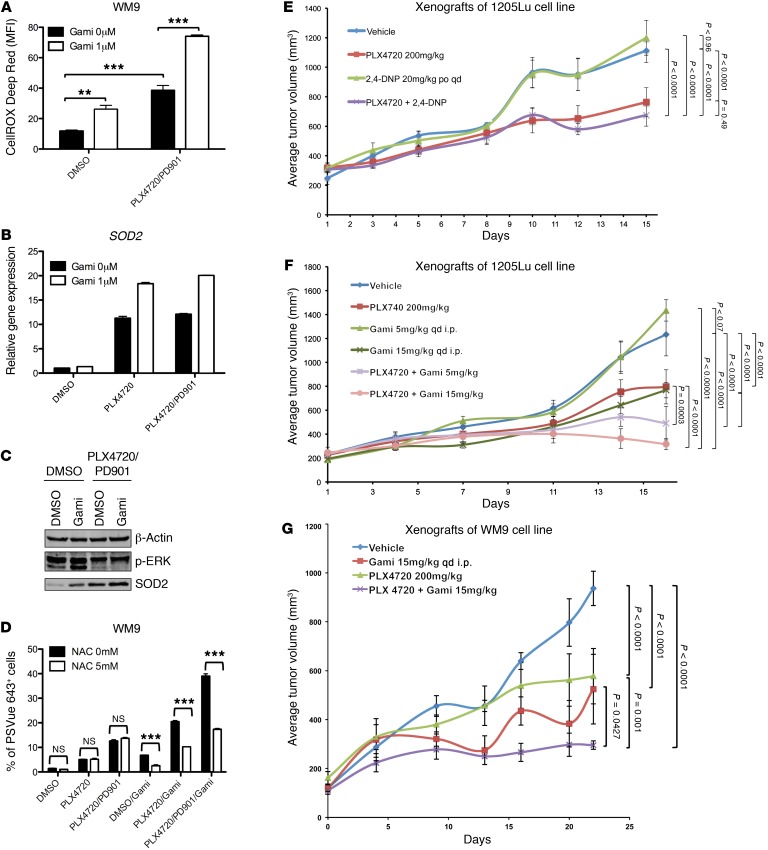
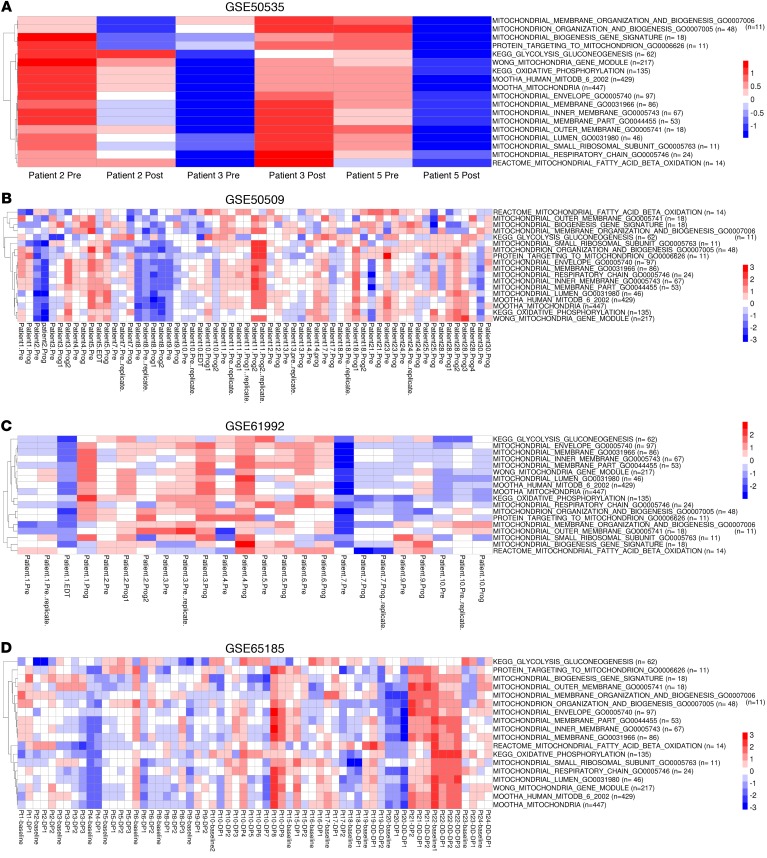
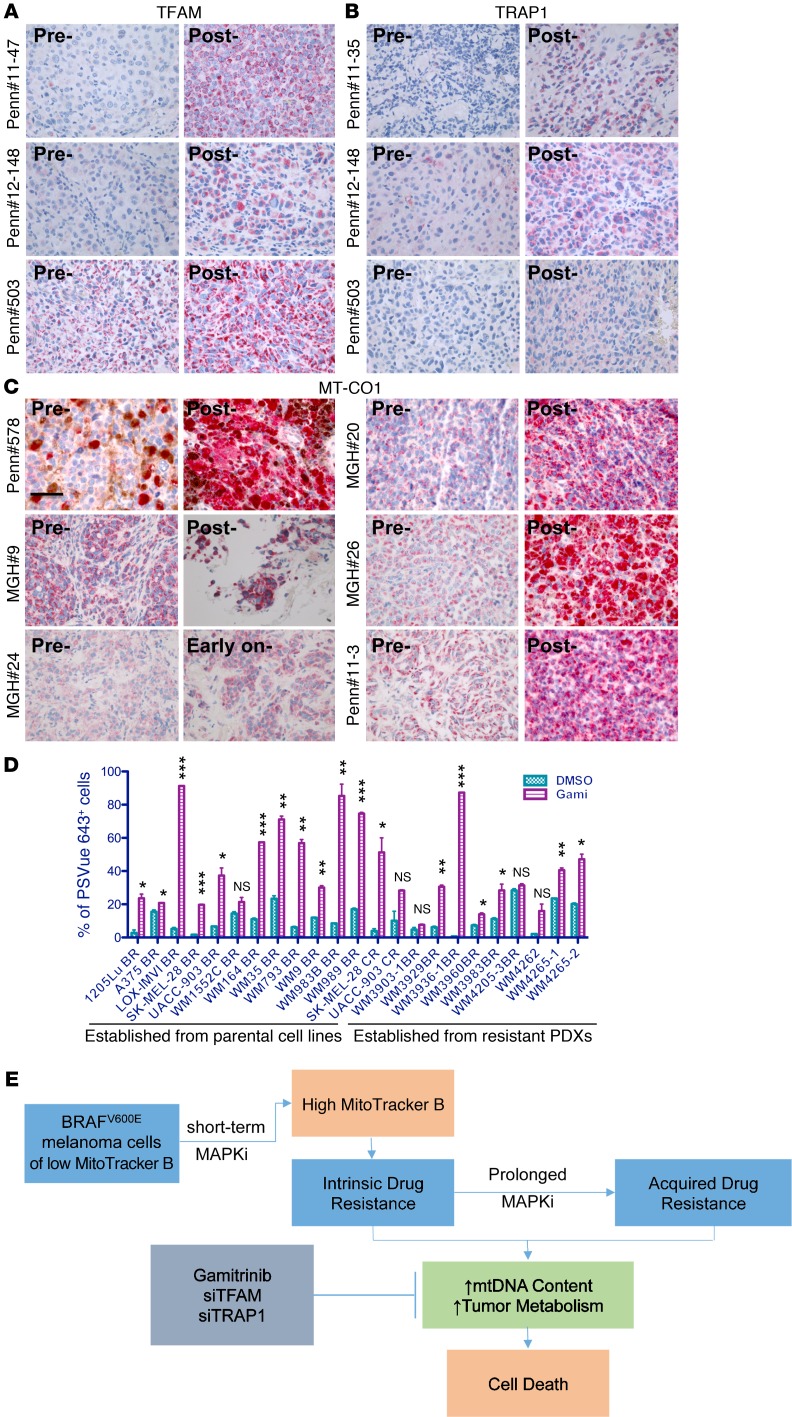
Targeting multiple components of the MAPK pathway can prolong the survival of patients with BRAFV600E melanoma. This approach is not curative, as some BRAF-mutated melanoma cells are intrinsically resistant to MAPK inhibitors (MAPKi). At the systemic level, our knowledge of how signaling pathways underlie drug resistance needs to be further expanded. Here, we have shown that intrinsically resistant BRAF-mutated melanoma cells with a low basal level of mitochondrial biogenesis depend on this process to survive MAPKi. Intrinsically resistant cells exploited an integrated stress response, exhibited an increase in mitochondrial DNA content, and required oxidative phosphorylation to meet their bioenergetic needs. We determined that intrinsically resistant cells rely on the genes encoding TFAM, which controls mitochondrial genome replication and transcription, and TRAP1, which regulates mitochondrial protein folding. Therefore, we targeted mitochondrial biogenesis with a mitochondrium-targeted, small-molecule HSP90 inhibitor (Gamitrinib), which eradicated intrinsically resistant cells and augmented the efficacy of MAPKi by inducing mitochondrial dysfunction and inhibiting tumor bioenergetics. A subset of tumor biopsies from patients with disease progression despite MAPKi treatment showed increased mitochondrial biogenesis and tumor bioenergetics. A subset of acquired drug-resistant melanoma cell lines was sensitive to Gamitrinib. Our study establishes mitochondrial biogenesis, coupled with aberrant tumor bioenergetics, as a potential therapy escape mechanism and paves the way for a rationale-based combinatorial strategy to improve the efficacy of MAPKi.
Figures

Comment in
-
Breaking BRAF(V600E)-drug resistance by stressing mitochondria.Pigment Cell Melanoma Res. 2016 Jul;29(4):401-3. doi: 10.1111/pcmr.12486. Pigment Cell Melanoma Res. 2016. PMID: 27135210 Free PMC article. No abstract available.
Similar articles
-
Tumor Necrosis Factor Receptor-associated Protein 1 (TRAP1) Mutation and TRAP1 Inhibitor Gamitrinib-triphenylphosphonium (G-TPP) Induce a Forkhead Box O (FOXO)-dependent Cell Protective Signal from Mitochondria.J Biol Chem. 2016 Jan 22;291(4):1841-1853. doi: 10.1074/jbc.M115.656934. Epub 2015 Dec 2. J Biol Chem. 2016. PMID: 26631731 Free PMC article.
-
A Novel Mitochondrial Inhibitor Blocks MAPK Pathway and Overcomes MAPK Inhibitor Resistance in Melanoma.Clin Cancer Res. 2019 Nov 1;25(21):6429-6442. doi: 10.1158/1078-0432.CCR-19-0836. Epub 2019 Aug 22. Clin Cancer Res. 2019. PMID: 31439581 Free PMC article.
-
A Fatty Acid Oxidation-dependent Metabolic Shift Regulates the Adaptation of BRAF-mutated Melanoma to MAPK Inhibitors.Clin Cancer Res. 2019 Nov 15;25(22):6852-6867. doi: 10.1158/1078-0432.CCR-19-0253. Epub 2019 Aug 2. Clin Cancer Res. 2019. PMID: 31375515 Free PMC article.
-
The Potential of Targeting P53 and HSP90 Overcoming Acquired MAPKi-Resistant Melanoma.Curr Treat Options Oncol. 2019 Feb 18;20(3):22. doi: 10.1007/s11864-019-0622-9. Curr Treat Options Oncol. 2019. PMID: 30778775 Review.
-
TRAP1 regulation of mitochondrial life or death decision in cancer cells and mitochondria-targeted TRAP1 inhibitors.BMB Rep. 2012 Jan;45(1):1-6. doi: 10.5483/bmbrep.2012.45.1.1. BMB Rep. 2012. PMID: 22281005 Review.
Cited by
-
Heterogeneity of Metabolic Vulnerability in Imatinib -Resistant Gastrointestinal Stromal Tumor.Cells. 2020 May 26;9(6):1333. doi: 10.3390/cells9061333. Cells. 2020. PMID: 32466502 Free PMC article.
-
Understanding the Intersections between Metabolism and Cancer Biology.Cell. 2017 Feb 9;168(4):657-669. doi: 10.1016/j.cell.2016.12.039. Cell. 2017. PMID: 28187287 Free PMC article. Review.
-
The Glutaminase Inhibitor CB-839 (Telaglenastat) Enhances the Antimelanoma Activity of T-Cell-Mediated Immunotherapies.Mol Cancer Ther. 2021 Mar;20(3):500-511. doi: 10.1158/1535-7163.MCT-20-0430. Epub 2020 Dec 23. Mol Cancer Ther. 2021. PMID: 33361272 Free PMC article.
-
p53 Affects PGC1α Stability Through AKT/GSK-3β to Enhance Cisplatin Sensitivity in Non-Small Cell Lung Cancer.Front Oncol. 2020 Aug 21;10:1252. doi: 10.3389/fonc.2020.01252. eCollection 2020. Front Oncol. 2020. PMID: 32974127 Free PMC article.
-
Mitochondria-targeted magnolol inhibits OXPHOS, proliferation, and tumor growth via modulation of energetics and autophagy in melanoma cells.Cancer Treat Res Commun. 2020;25:100210. doi: 10.1016/j.ctarc.2020.100210. Epub 2020 Sep 17. Cancer Treat Res Commun. 2020. PMID: 32987287 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
