

A role for FOXO1 in BCR-ABL1-independent tyrosine kinase inhibitor resistance in chronic myeloid leukemia
- PMID: 27044711
- PMCID: PMC4935980
- DOI: 10.1038/leu.2016.51
A role for FOXO1 in BCR-ABL1-independent tyrosine kinase inhibitor resistance in chronic myeloid leukemia
Abstract
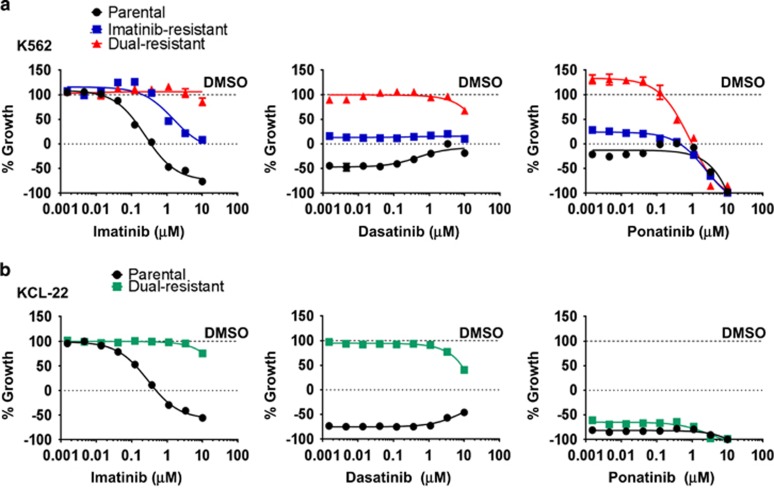
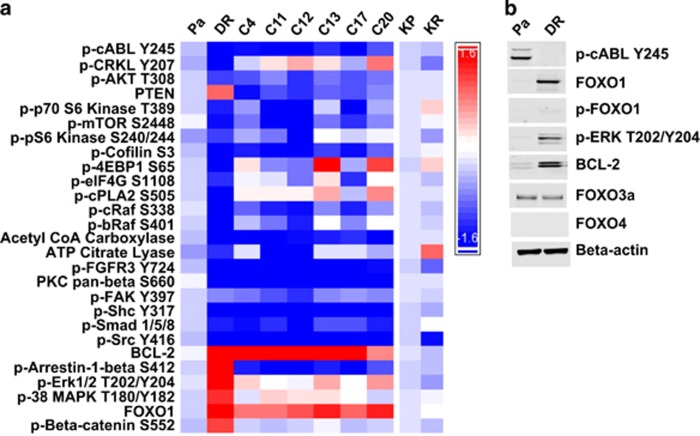
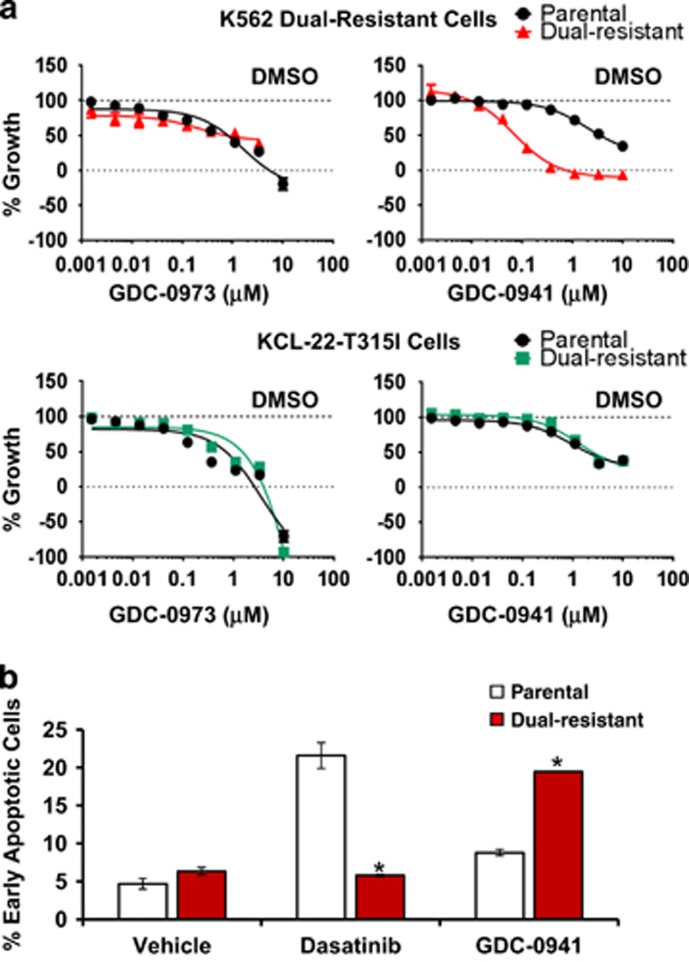
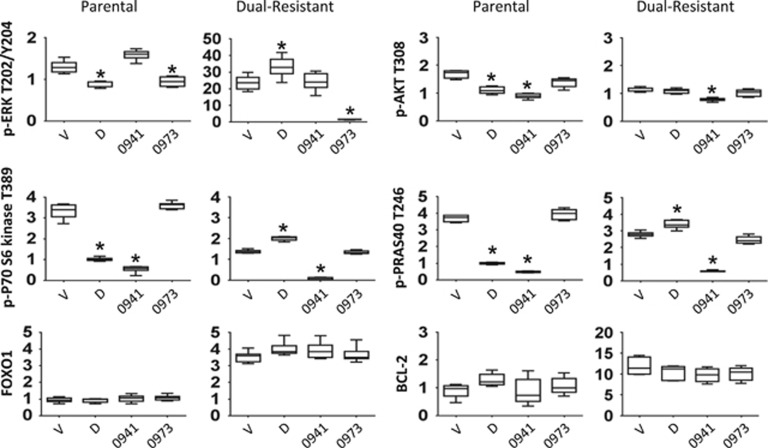
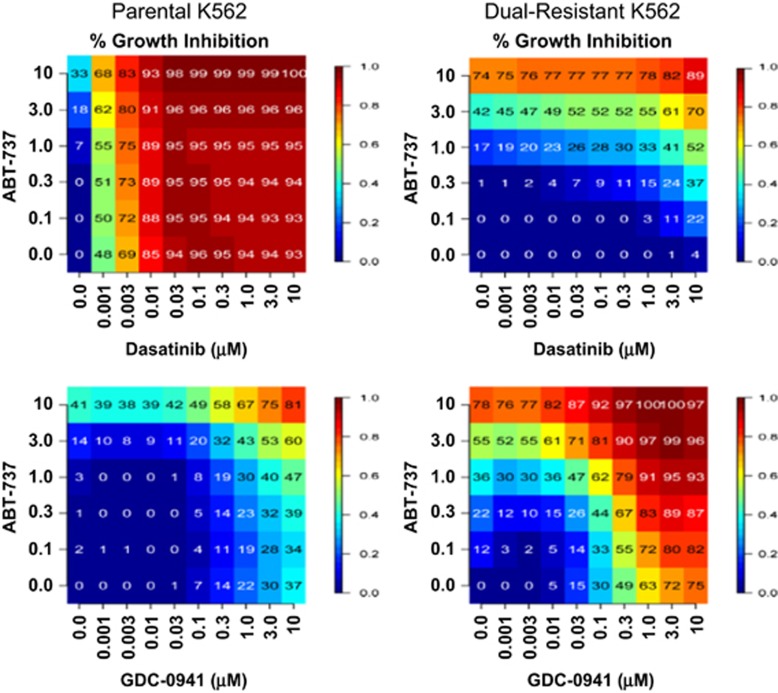
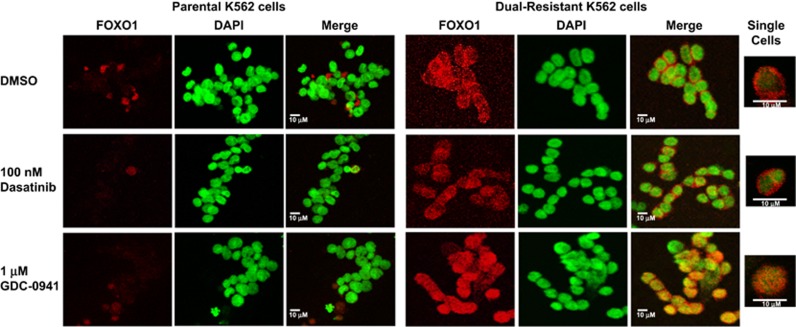
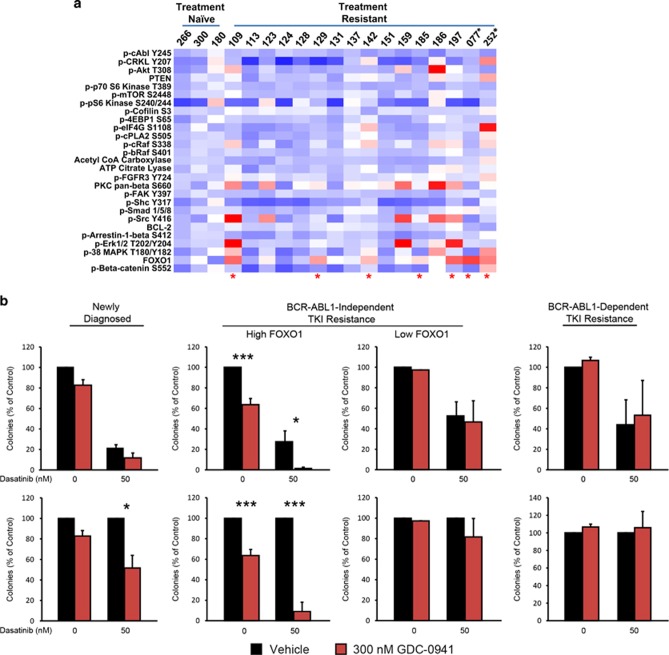
Chronic myeloid leukemia (CML) patients who relapse on imatinib due to acquired ABL1 kinase domain mutations are successfully treated with second-generation ABL1-tyrosine kinase inhibitors (ABL-TKIs) such as dasatinib, nilotinib or ponatinib. However, ~40% of relapsed patients have uncharacterized BCR-ABL1 kinase-independent mechanisms of resistance. To identify these mechanisms of resistance and potential treatment options, we generated ABL-TKI-resistant K562 cells through prolonged sequential exposure to imatinib and dasatinib. Dual-resistant K562 cells lacked BCR-ABL1 kinase domain mutations, but acquired other genomic aberrations that were characterized by next-generation sequencing and copy number analyses. Proteomics showed that dual-resistant cells had elevated levels of FOXO1, phospho-ERK and BCL-2, and that dasatinib no longer inhibited substrates of the PI3K/AKT pathway. In contrast to parental cells, resistant cells were sensitive to growth inhibition and apoptosis induced by the class I PI3K inhibitor, GDC-0941 (pictilisib), which also induced FOXO1 nuclear translocation. FOXO1 was elevated in a subset of primary specimens from relapsed CML patients lacking BCR-ABL1 kinase domain mutations, and these samples were responsive to GDC-0941 treatment ex vivo. We conclude that elevated FOXO1 contributes to BCR-ABL1 kinase-independent resistance experienced by these CML patients and that PI3K inhibition coupled with BCR-ABL1 inhibition may represent a novel therapeutic approach.
Conflict of interest statement
Some of the authors are employees of Roche/Genentech. We used our in-house molecules, GDC-0941 and GDC-0973, as representative PI3K and MEK kinase inhibitors, respectively. These molecules have been published and are available from commercial sources. Furthermore, our findings should be applicable to similar inhibitors from other sources. The remaining authors declare no conflict of interest.
Figures
Similar articles
-
BCR-ABL1-independent PI3Kinase activation causing imatinib-resistance.J Hematol Oncol. 2011 Feb 7;4:6. doi: 10.1186/1756-8722-4-6. J Hematol Oncol. 2011. PMID: 21299849 Free PMC article.
-
Contributions of MET activation to BCR-ABL1 tyrosine kinase inhibitor resistance in chronic myeloid leukemia cells.Oncotarget. 2017 Jun 13;8(24):38717-38730. doi: 10.18632/oncotarget.16314. Oncotarget. 2017. PMID: 28418880 Free PMC article.
-
Cotreatment with vorinostat (suberoylanilide hydroxamic acid) enhances activity of dasatinib (BMS-354825) against imatinib mesylate-sensitive or imatinib mesylate-resistant chronic myelogenous leukemia cells.Clin Cancer Res. 2006 Oct 1;12(19):5869-78. doi: 10.1158/1078-0432.CCR-06-0980. Clin Cancer Res. 2006. PMID: 17020995
-
[MET/ERK and MET/JNK Pathway Activation Is Involved in BCR-ABL Inhibitor-resistance in Chronic Myeloid Leukemia].Yakugaku Zasshi. 2018;138(12):1461-1466. doi: 10.1248/yakushi.18-00142. Yakugaku Zasshi. 2018. PMID: 30504658 Review. Japanese.
-
BCR-ABL tyrosine kinase inhibitors in the treatment of Philadelphia chromosome positive chronic myeloid leukemia: a review.Leuk Res. 2010 Oct;34(10):1255-68. doi: 10.1016/j.leukres.2010.04.016. Leuk Res. 2010. PMID: 20537386 Review.
Cited by
-
Mechanisms of Disease Progression and Resistance to Tyrosine Kinase Inhibitor Therapy in Chronic Myeloid Leukemia: An Update.Int J Mol Sci. 2019 Dec 5;20(24):6141. doi: 10.3390/ijms20246141. Int J Mol Sci. 2019. PMID: 31817512 Free PMC article. Review.
-
SRSF1 mediates cytokine-induced impaired imatinib sensitivity in chronic myeloid leukemia.Leukemia. 2020 Jul;34(7):1787-1798. doi: 10.1038/s41375-020-0732-1. Epub 2020 Feb 12. Leukemia. 2020. PMID: 32051529 Free PMC article.
-
Targeting PFKFB3 sensitizes chronic myelogenous leukemia cells to tyrosine kinase inhibitor.Oncogene. 2018 May;37(21):2837-2849. doi: 10.1038/s41388-018-0157-8. Epub 2018 Mar 7. Oncogene. 2018. PMID: 29511345
-
Insights into existing and futuristic treatment approach for chronic myeloid leukaemia.Indian J Med Res. 2024 May;159(5):455-467. doi: 10.25259/ijmr_1716_22. Indian J Med Res. 2024. PMID: 39382408 Free PMC article. Review.
-
Compact CRISPR genetic screens enabled by improved guide RNA library cloning.Genome Biol. 2024 Jan 19;25(1):25. doi: 10.1186/s13059-023-03132-3. Genome Biol. 2024. PMID: 38243310 Free PMC article.
References
-
- O'Hare T, Zabriskie MS, Eiring AM, Deininger MW. Pushing the limits of targeted therapy in chronic myeloid leukaemia. Nat Rev Cancer 2012; 12: 513–526. - PubMed
-
- Hochhaus A, Kreil S, Corbin AS, La Rosee P, Muller MC, Lahaye T et al. Molecular and chromosomal mechanisms of resistance to imatinib (STI571) therapy. Leukemia 2002; 16: 2190–2196. - PubMed
-
- Soverini S, Colarossi S, Gnani A, Rosti G, Castagnetti F, Poerio A et al. Contribution of ABL kinase domain mutations to imatinib resistance in different subsets of Philadelphia positive patients: by the GIMEMA Working Party on Chronic Myeloid Leukemia. Clin Cancer Res 2006; 12: 7374–7379. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
