

Genic insights from integrated human proteomics in GeneCards
- PMID: 27048349
- PMCID: PMC4820835
- DOI: 10.1093/database/baw030
Genic insights from integrated human proteomics in GeneCards
Abstract
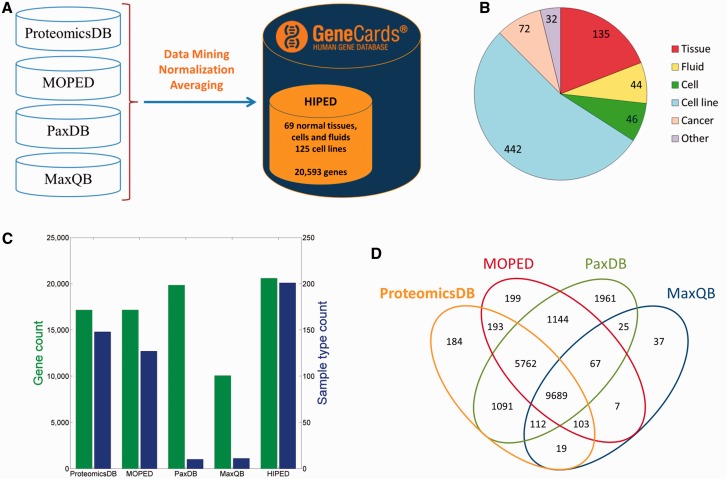
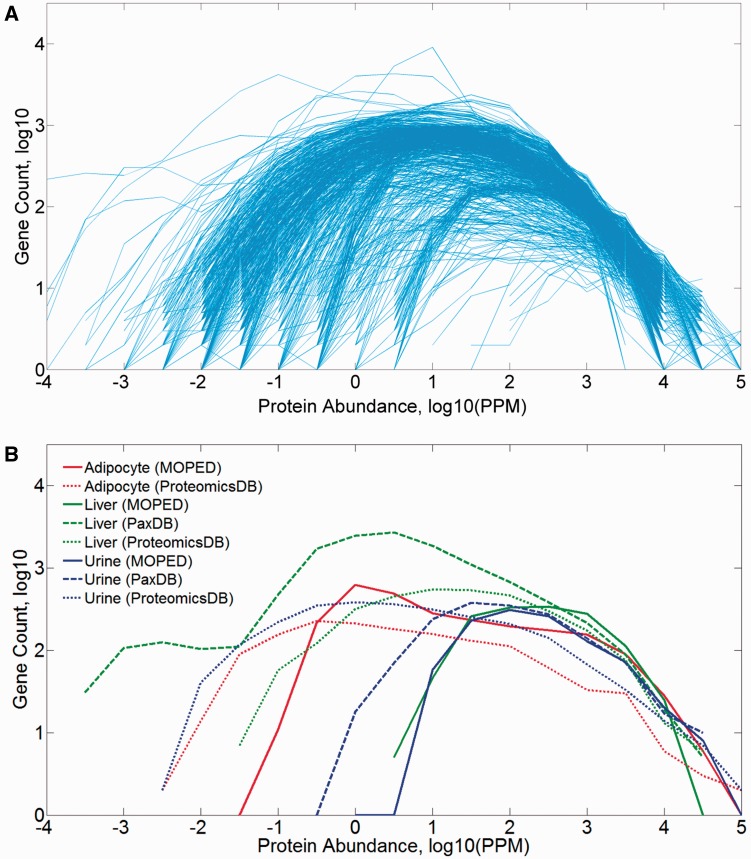
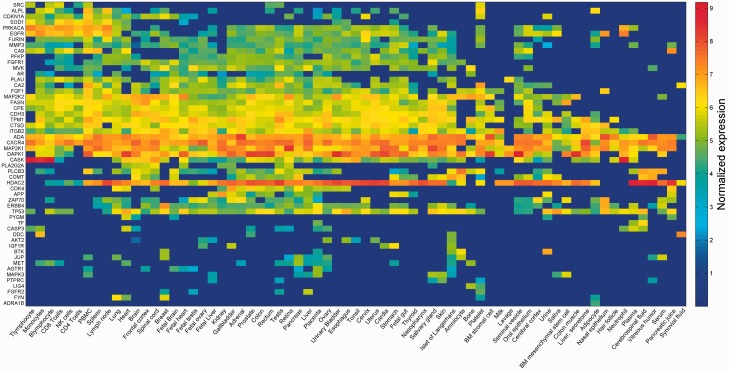
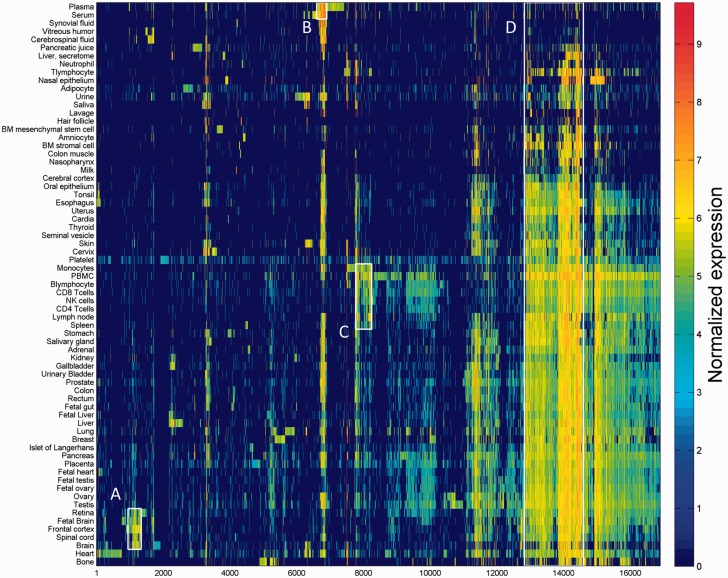
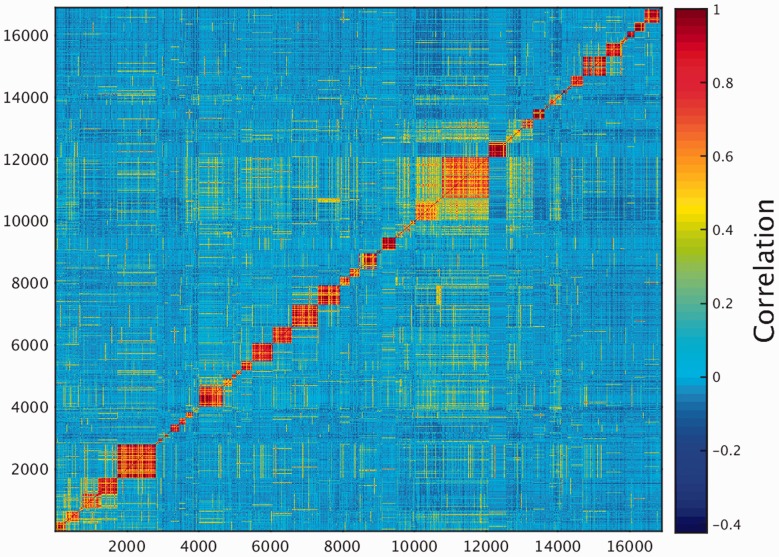
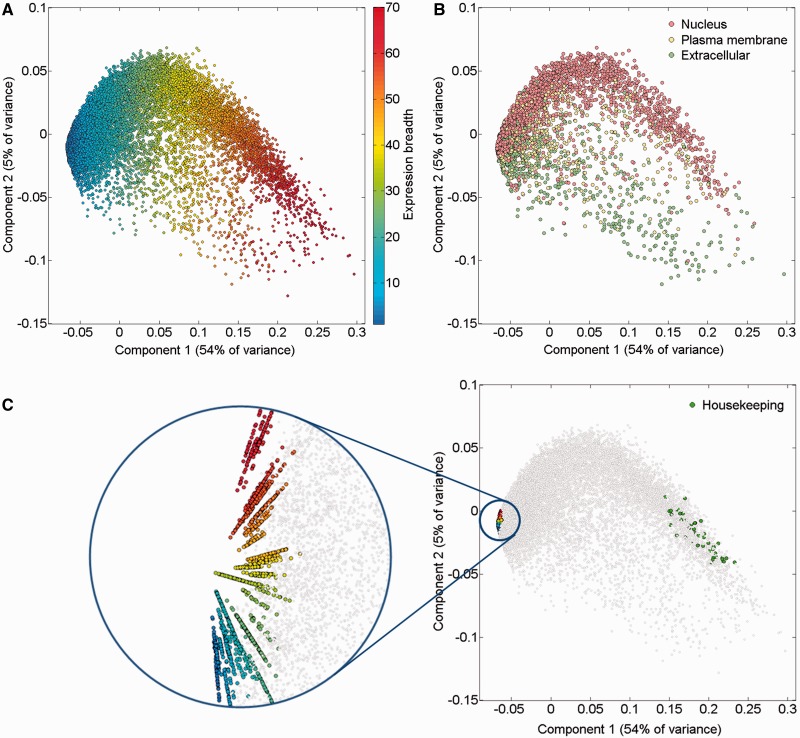
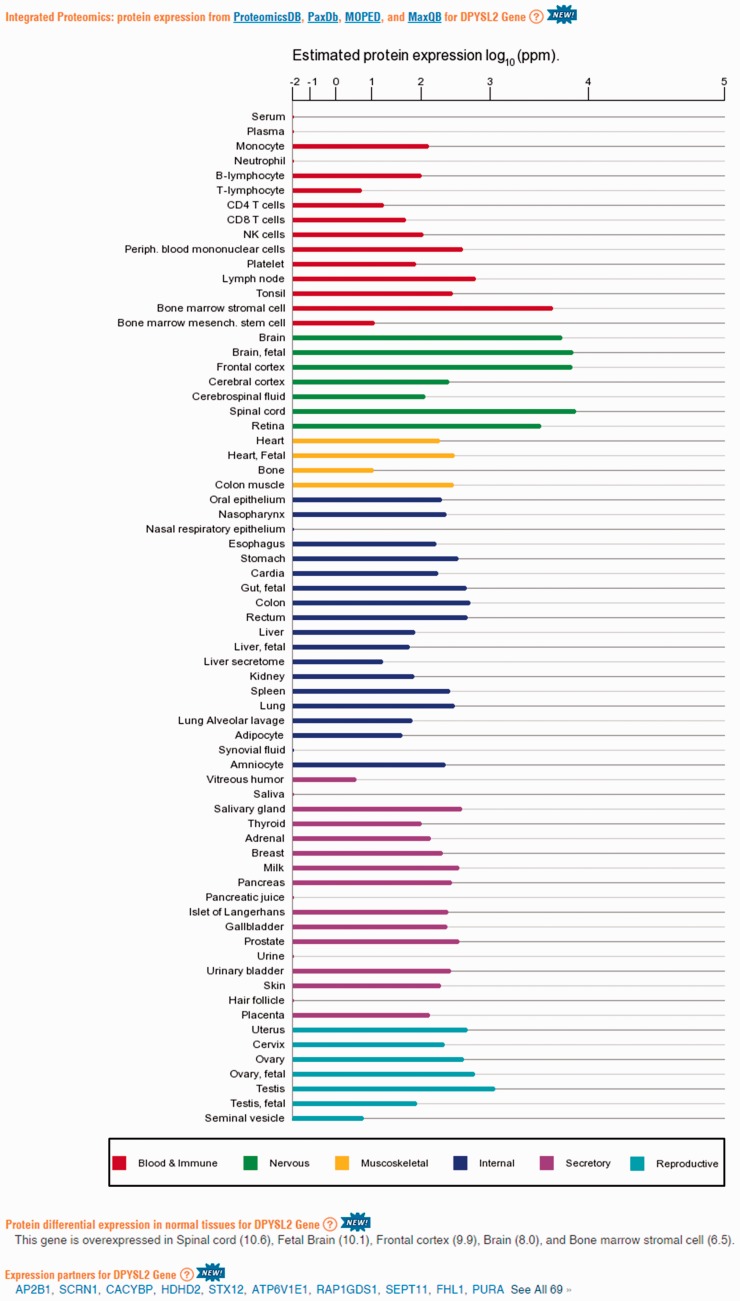
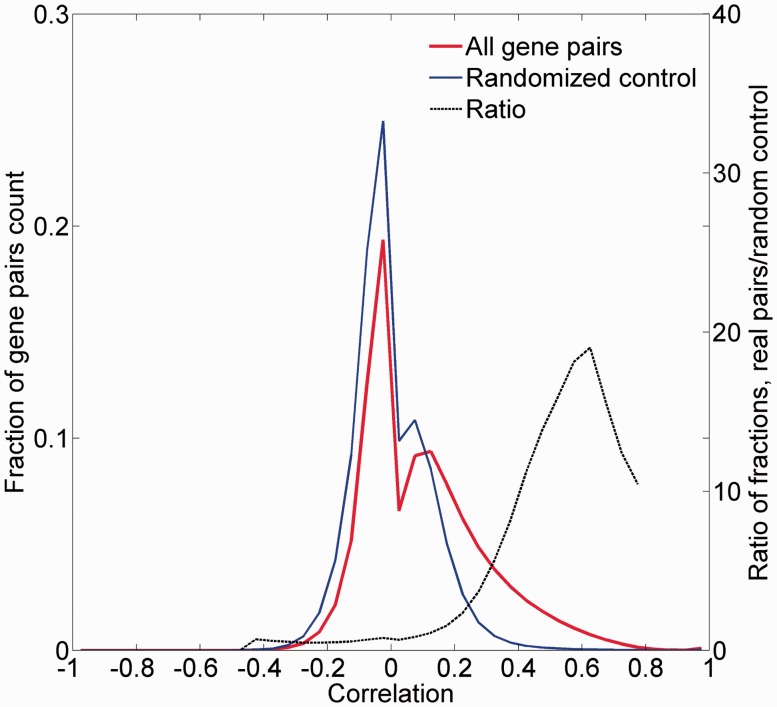
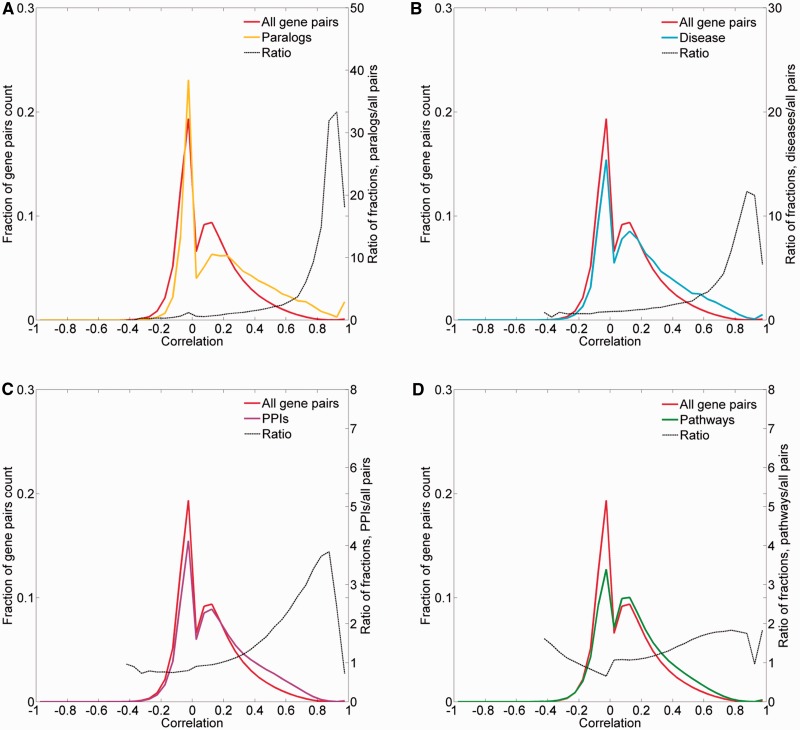
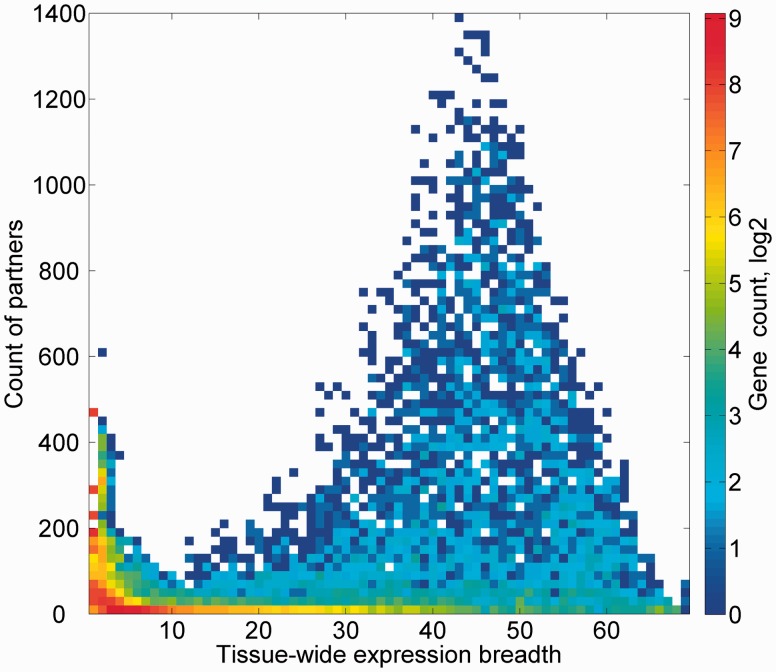
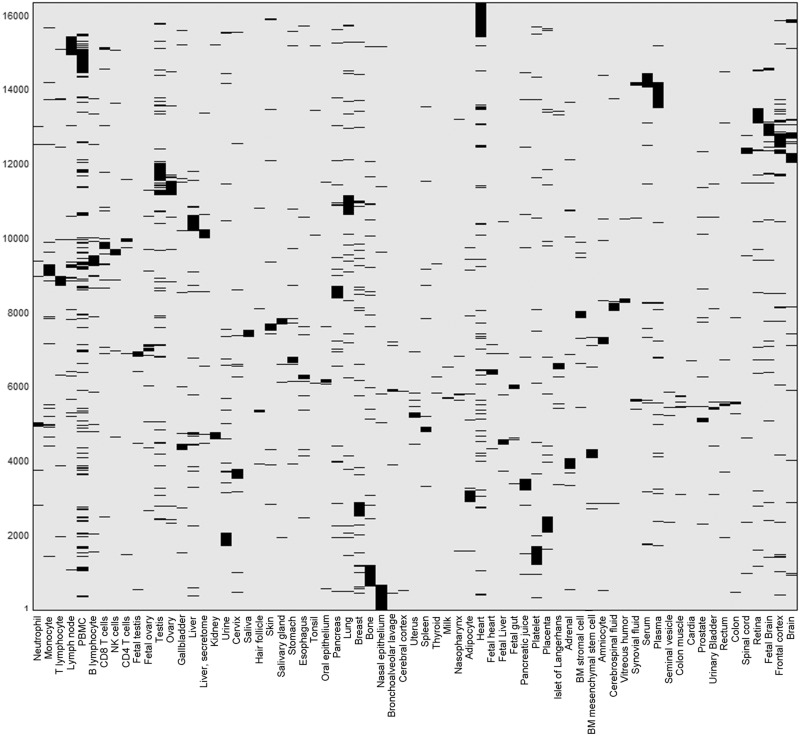
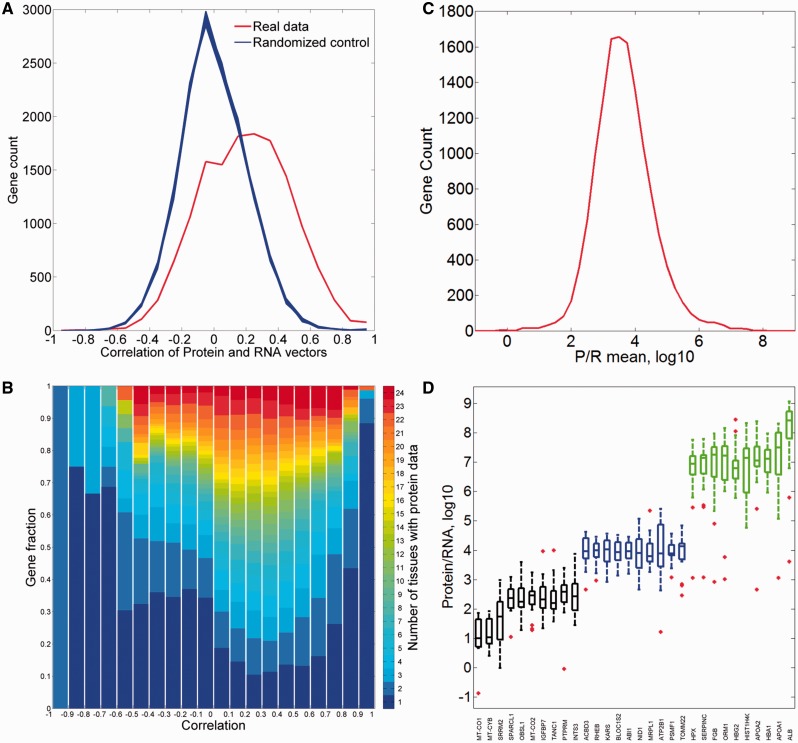
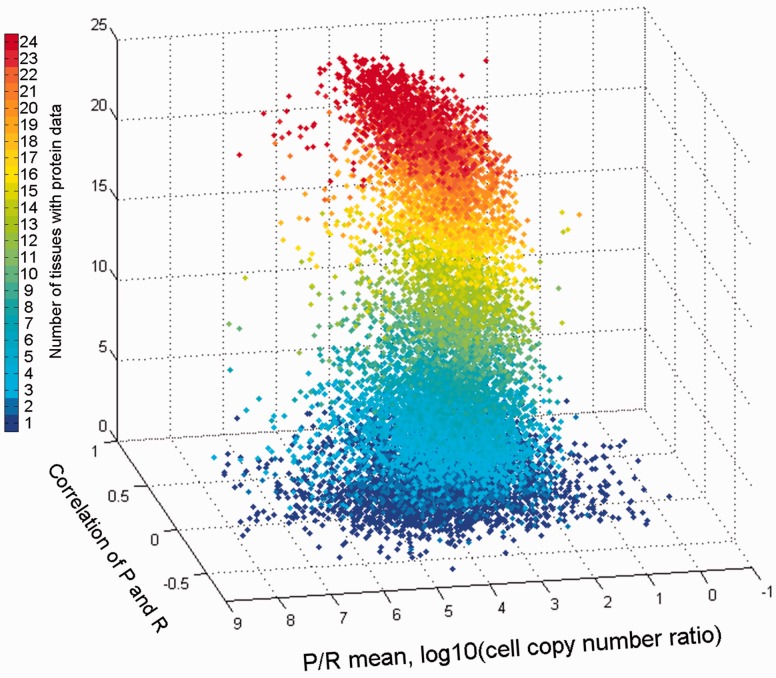
GeneCards is a one-stop shop for searchable human gene annotations (http://www.genecards.org/). Data are automatically mined from ∼120 sources and presented in an integrated web card for every human gene. We report the application of recent advances in proteomics to enhance gene annotation and classification in GeneCards. First, we constructed the Human Integrated Protein Expression Database (HIPED), a unified database of protein abundance in human tissues, based on the publically available mass spectrometry (MS)-based proteomics sources ProteomicsDB, Multi-Omics Profiling Expression Database, Protein Abundance Across Organisms and The MaxQuant DataBase. The integrated database, residing within GeneCards, compares favourably with its individual sources, covering nearly 90% of human protein-coding genes. For gene annotation and comparisons, we first defined a protein expression vector for each gene, based on normalized abundances in 69 normal human tissues. This vector is portrayed in the GeneCards expression section as a bar graph, allowing visual inspection and comparison. These data are juxtaposed with transcriptome bar graphs. Using the protein expression vectors, we further defined a pairwise metric that helps assess expression-based pairwise proximity. This new metric for finding functional partners complements eight others, including sharing of pathways, gene ontology (GO) terms and domains, implemented in the GeneCards Suite. In parallel, we calculated proteome-based differential expression, highlighting a subset of tissues that overexpress a gene and subserving gene classification. This textual annotation allows users of VarElect, the suite's next-generation phenotyper, to more effectively discover causative disease variants. Finally, we define the protein-RNA expression ratio and correlation as yet another attribute of every gene in each tissue, adding further annotative information. The results constitute a significant enhancement of several GeneCards sections and help promote and organize the genome-wide structural and functional knowledge of the human proteome. Database URL:http://www.genecards.org/.
© The Author(s) 2016. Published by Oxford University Press.
Figures
References
-
- Mann M., Kulak N.A., Nagaraj N. et al. (2013) The coming age of complete, accurate, and ubiquitous proteomes. Mol. Cell, 49, 583–590. - PubMed
-
- Paik Y.K., Jeong S.K., Omenn G.S. et al. (2012) The Chromosome-Centric Human Proteome Project for cataloging proteins encoded in the genome. Nat. Biotechnol., 30, 221–223. - PubMed
-
- Wilhelm M., Schlegl J., Hahne H. et al. (2014) Mass-spectrometry-based draft of the human proteome. Nature, 509, 582–587. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
