

Necrosome core machinery: MLKL
- PMID: 27048809
- PMCID: PMC11108342
- DOI: 10.1007/s00018-016-2190-5
Necrosome core machinery: MLKL
Abstract
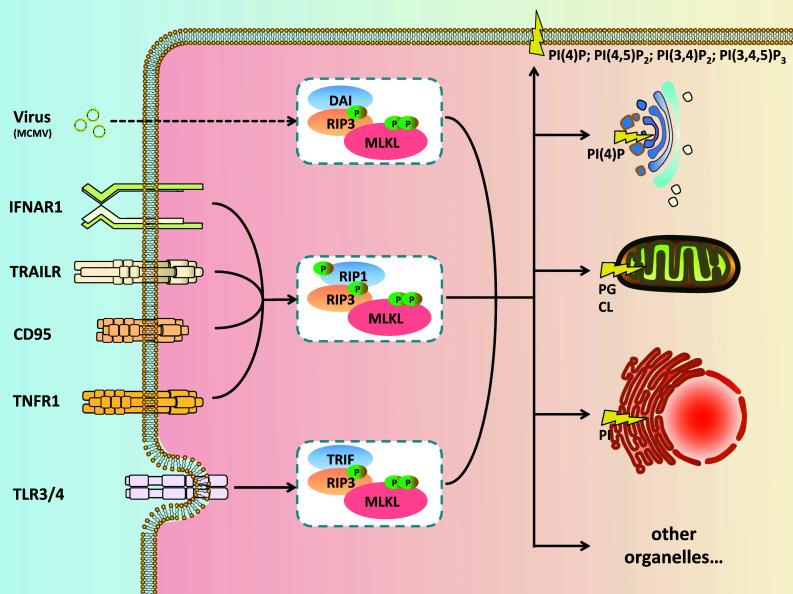
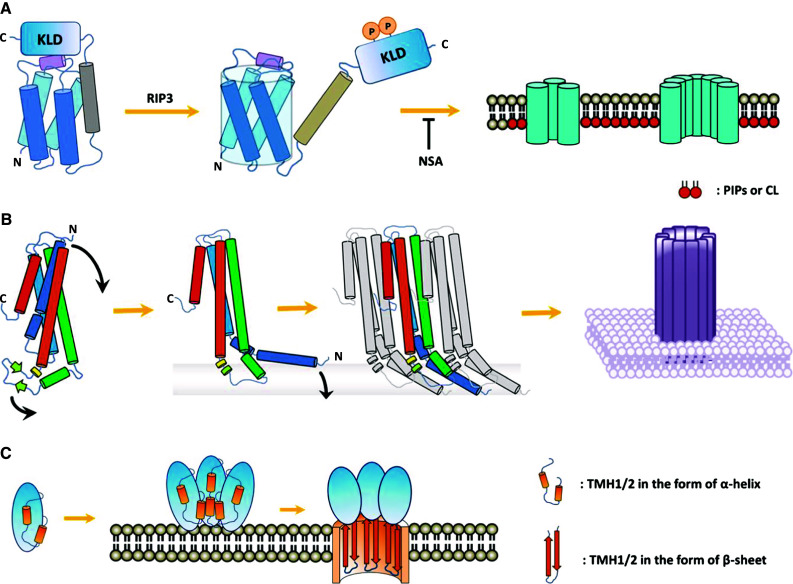
In the study of regulated cell death, the rapidly expanding field of regulated necrosis, in particular necroptosis, has been drawing much attention. The signaling of necroptosis represents a sophisticated form of a death pathway. Anti-caspase mechanisms (e.g., using inhibitors of caspases, or genetic ablation of caspase-8) switch cell fate from apoptosis to necroptosis. The initial extracellular death signals regulate RIP1 and RIP3 kinase activation. The RIP3-associated death complex assembly is necessary and sufficient to initiate necroptosis. MLKL was initially identified as an essential mediator of RIP1/RIP3 kinase-initiated necroptosis. Recent studies on the signal transduction using chemical tools and biomarkers support the idea that MLKL is able to make more functional sense for the core machinery of the necroptosis death complex, called the necrosome, to connect to the necroptosis execution. The experimental data available now have pointed that the activated MLKL forms membrane-disrupting pores causing membrane leakage, which extends the prototypical concept of morphological and biochemical events following necroptosis happening in vivo. The key role of MLKL in necroptosis signaling thus sheds light on the logic underlying this unique "membrane-explosive" cell death pathway. In this review, we provide the general concepts and strategies that underlie signal transduction of this form of cell death, and then focus specifically on the role of MLKL in necroptosis.
Keywords: MLKL; Necroptosis; Necrosome; Pore-forming protein; Regulated cell death.
Figures
Similar articles
-
PI3K mediates tumor necrosis factor induced-necroptosis through initiating RIP1-RIP3-MLKL signaling pathway activation.Cytokine. 2020 May;129:155046. doi: 10.1016/j.cyto.2020.155046. Epub 2020 Feb 28. Cytokine. 2020. PMID: 32114297
-
RIP1, RIP3, and MLKL Contribute to Cell Death Caused by Clostridium perfringens Enterotoxin.mBio. 2019 Dec 17;10(6):e02985-19. doi: 10.1128/mBio.02985-19. mBio. 2019. PMID: 31848291 Free PMC article.
-
Diverse sequence determinants control human and mouse receptor interacting protein 3 (RIP3) and mixed lineage kinase domain-like (MLKL) interaction in necroptotic signaling.J Biol Chem. 2013 Jun 7;288(23):16247-16261. doi: 10.1074/jbc.M112.435545. Epub 2013 Apr 23. J Biol Chem. 2013. PMID: 23612963 Free PMC article.
-
Biomarkers for the detection of necroptosis.Cell Mol Life Sci. 2016 Jun;73(11-12):2177-81. doi: 10.1007/s00018-016-2192-3. Epub 2016 Apr 11. Cell Mol Life Sci. 2016. PMID: 27066893 Free PMC article. Review.
-
Necroptosis in health and diseases.Semin Cell Dev Biol. 2014 Nov;35:14-23. doi: 10.1016/j.semcdb.2014.07.013. Epub 2014 Aug 1. Semin Cell Dev Biol. 2014. PMID: 25087983 Review.
Cited by
-
Necroptosis and Alzheimer's Disease: Pathogenic Mechanisms and Therapeutic Opportunities.J Alzheimers Dis. 2023;94(s1):S367-S386. doi: 10.3233/JAD-220809. J Alzheimers Dis. 2023. PMID: 36463451 Free PMC article. Review.
-
Emerging Role of ZBP1 in Z-RNA Sensing, Influenza Virus-Induced Cell Death, and Pulmonary Inflammation.mBio. 2022 Jun 28;13(3):e0040122. doi: 10.1128/mbio.00401-22. Epub 2022 May 19. mBio. 2022. PMID: 35587190 Free PMC article. Review.
-
Postconditioning with inhaled hydrogen attenuates skin ischemia/reperfusion injury through the RIP-MLKL-PGAM5/Drp1 necrotic pathway.Am J Transl Res. 2019 Jan 15;11(1):499-508. eCollection 2019. Am J Transl Res. 2019. PMID: 30788005 Free PMC article.
-
Drp1 and RB interaction to mediate mitochondria-dependent necroptosis induced by cadmium in hepatocytes.Cell Death Dis. 2019 Jul 8;10(7):523. doi: 10.1038/s41419-019-1730-y. Cell Death Dis. 2019. PMID: 31285421 Free PMC article.
-
TRADD Mediates RIPK1-Independent Necroptosis Induced by Tumor Necrosis Factor.Front Cell Dev Biol. 2020 Jan 22;7:393. doi: 10.3389/fcell.2019.00393. eCollection 2019. Front Cell Dev Biol. 2020. PMID: 32039207 Free PMC article.
References
-
- Vercammen D, Beyaert R, Denecker G, Goossens V, Van Loo G, Declercq W, Grooten J, Fiers W, Vandenabeele P. Inhibition of caspases increases the sensitivity of L929 cells to necrosis mediated by tumor necrosis factor. J Exp Med. 1998;187(9):1477–1485. doi: 10.1084/jem.187.9.1477. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
