

A Novel Plant Sesquiterpene Lactone Derivative, DETD-35, Suppresses BRAFV600E Mutant Melanoma Growth and Overcomes Acquired Vemurafenib Resistance in Mice
- PMID: 27048951
- PMCID: PMC4893919
- DOI: 10.1158/1535-7163.MCT-15-0973
A Novel Plant Sesquiterpene Lactone Derivative, DETD-35, Suppresses BRAFV600E Mutant Melanoma Growth and Overcomes Acquired Vemurafenib Resistance in Mice
Abstract
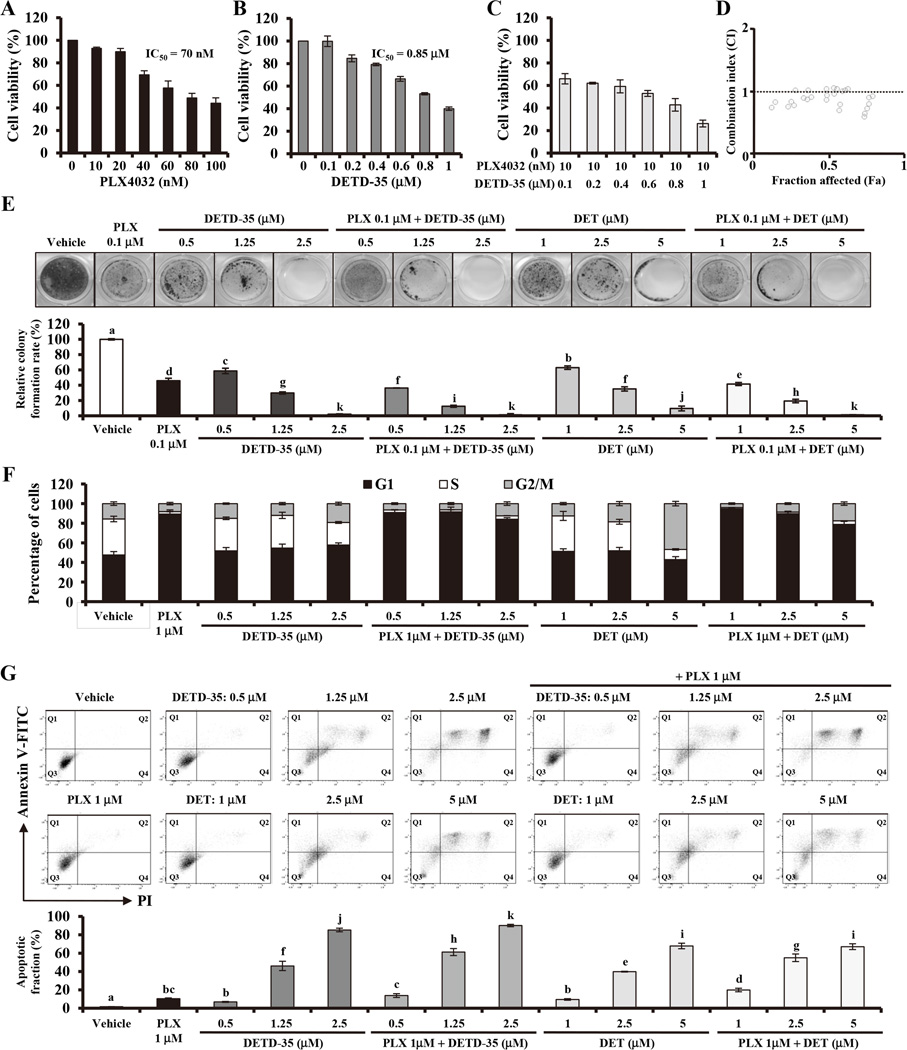
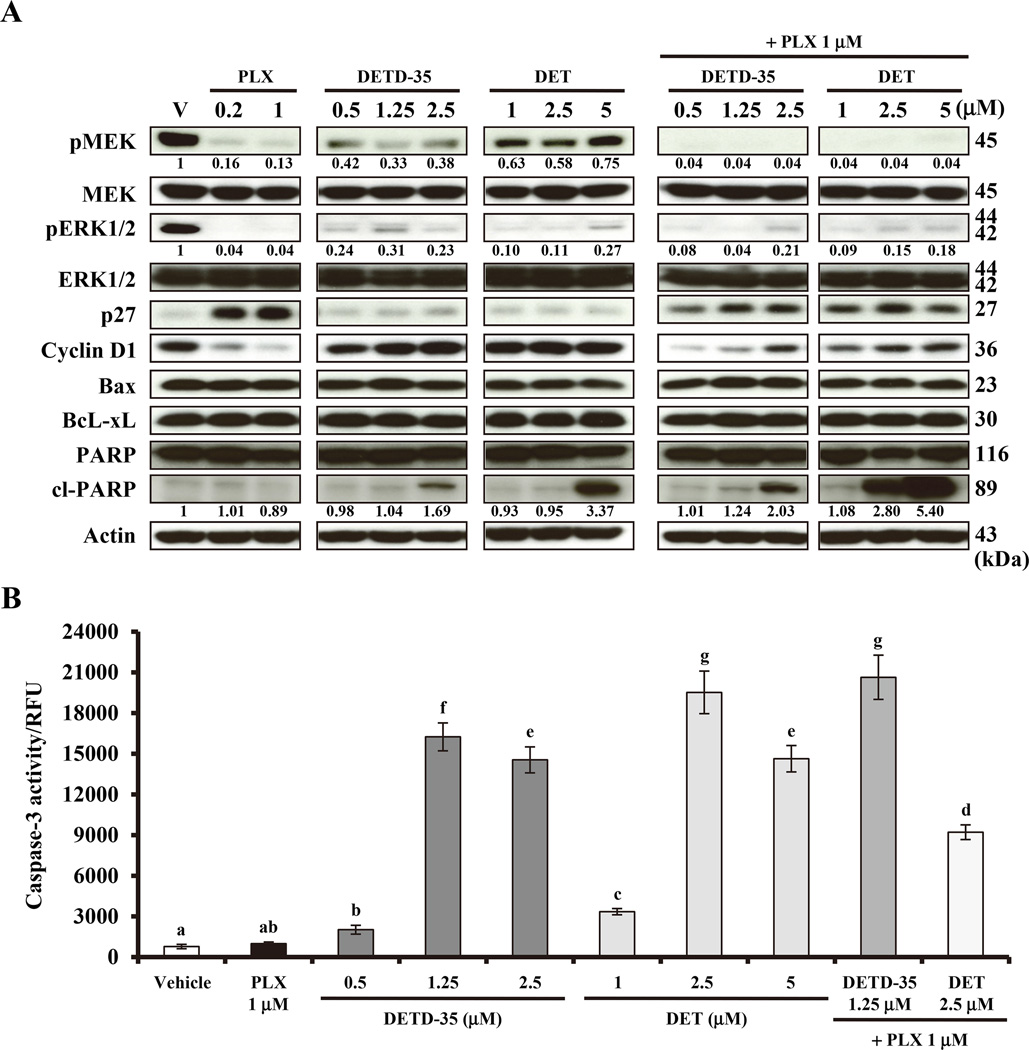
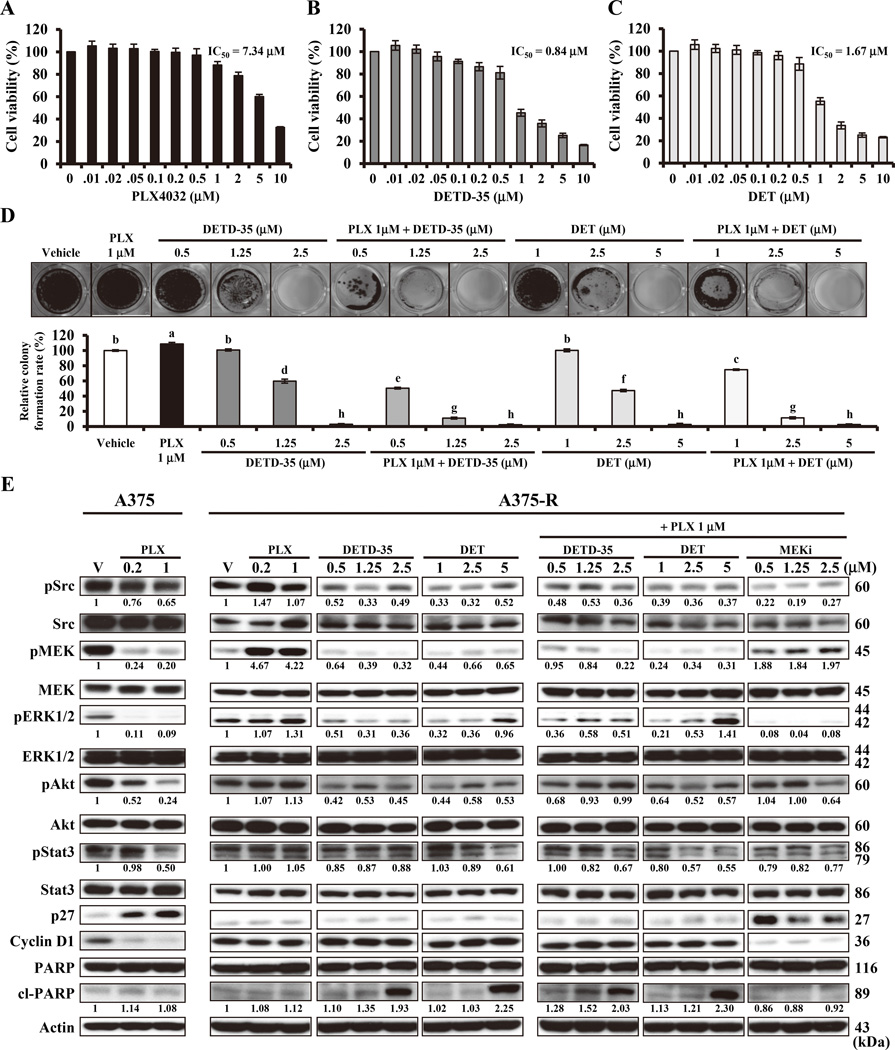
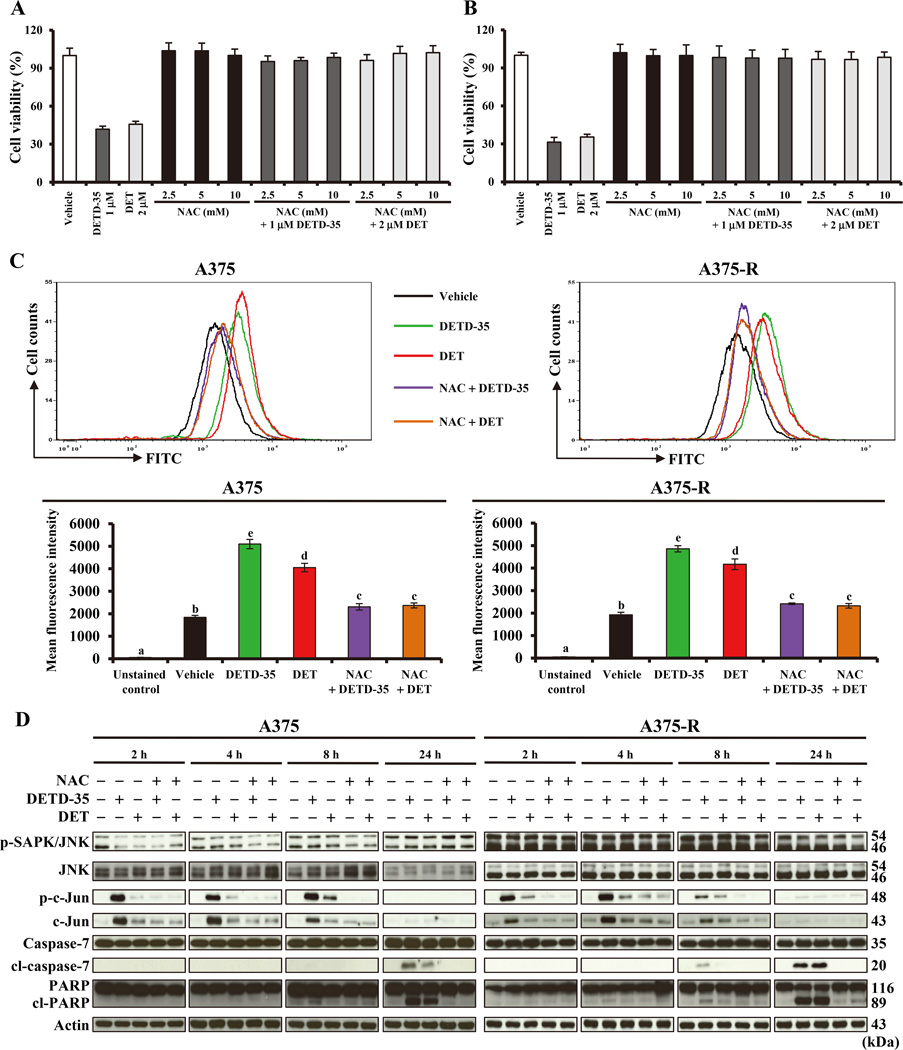
Acquired resistance to vemurafenib develops through reactivation of RAF/MEK/ERK signaling or bypass mechanisms. Recent combination therapies such as a MEK inhibitor combined with vemurafenib show improvement in major clinical end points, but the percentage of patients with adverse toxic events is higher than with vemurafenib monotherapy and most patients ultimately relapse. Therefore, there is an urgent need to develop new antimelanoma drugs and/or adjuvant agents for vemurafenib therapy. In this study, we created a novel semiorganically modified derivative, DETD-35, from deoxyelephantopin (DET), a plant sesquiterpene lactone demonstrated as an anti-inflammatory and anti-mammary tumor agent. Our results show that DETD-35 inhibited proliferation of a panel of melanoma cell lines, including acquired vemurafenib resistance A375 cells (A375-R) established in this study, with superior activities to DET and no cytotoxicity to normal melanocytes. DETD-35 suppressed tumor growth and reduced tumor mass as effectively as vemurafenib in A375 xenograft study. Furthermore, DETD-35 also reduced tumor growth in both acquired (A375-R) and intrinsic (A2058) vemurafenib resistance xenograft models, where vemurafenib showed no antitumor activity. Notably, the combination of DETD-35 and vemurafenib exhibited the most significant effects in both in vitro and in vivo xenograft studies due to synergism of the compound and the drug. Mechanistic studies suggested that DETD-35 overcame acquired vemurafenib resistance at least in part through deregulating MEK-ERK, Akt, and STAT3 signaling pathways and promoting apoptosis of cancer cells. Overall, our results suggest that DETD-35 may be useful as a therapeutic or adjuvant agent against BRAF(V600E) mutant and acquired vemurafenib resistance melanoma. Mol Cancer Ther; 15(6); 1163-76. ©2016 AACR.
©2016 American Association for Cancer Research.
Conflict of interest statement
The authors declare no conflict of interests.
Figures


References
-
- Greenlee RT, Hill-Harmon MB, Murray T, Thun M. Cancer statistics, 2001. CA Cancer J Clin. 2001;51:15–36. - PubMed
-
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65:5–29. - PubMed
-
- Halilovic E, Solit DB. Therapeutic strategies for inhibiting oncogenic BRAF signaling. Curr Opin Pharmacol. 2008;8:419–426. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
