

Lipotoxic lethal and sublethal stress signaling in hepatocytes: relevance to NASH pathogenesis
- PMID: 27049024
- PMCID: PMC5036373
- DOI: 10.1194/jlr.R066357
Lipotoxic lethal and sublethal stress signaling in hepatocytes: relevance to NASH pathogenesis
Erratum in
-
ERRATUM.J Lipid Res. 2017 Jan;58(1):299. doi: 10.1194/jlr.R066357ERR. J Lipid Res. 2017. PMID: 28042122 Free PMC article. No abstract available.
Abstract
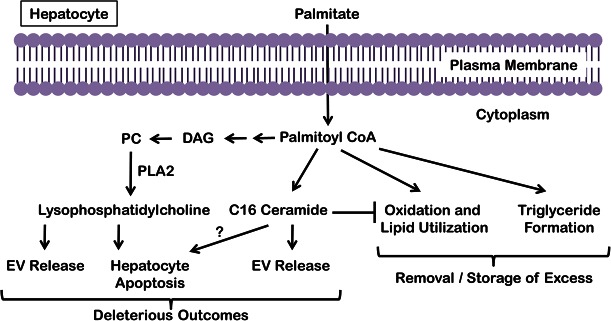
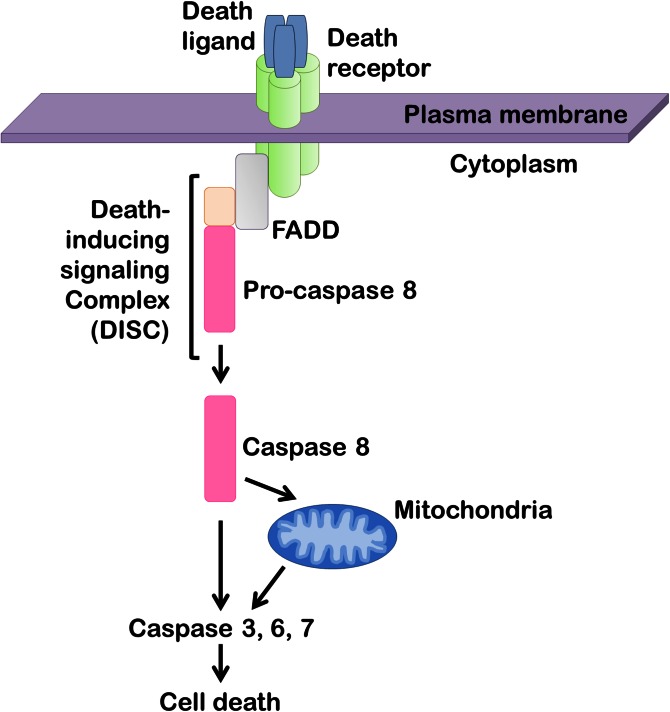
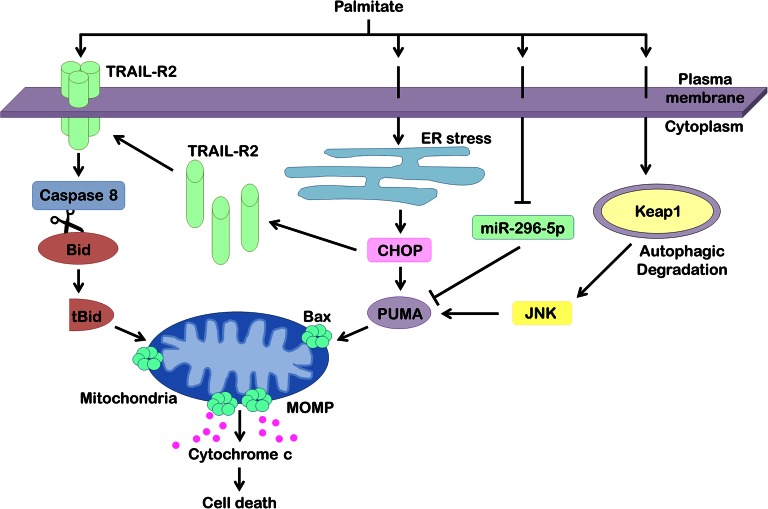
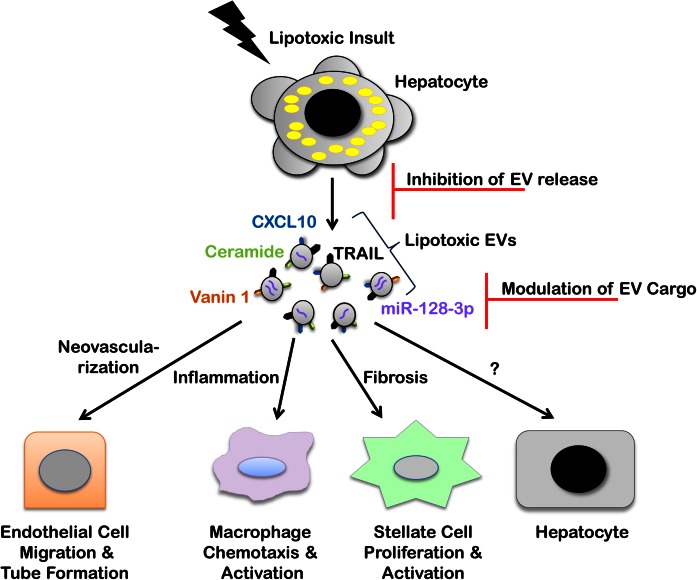
The accumulation of lipids is a histologic and biochemical hallmark of obesity-associated nonalcoholic fatty liver disease (NAFLD). A subset of NALFD patients develops progressive liver disease, termed nonalcoholic steatohepatitis, which is characterized by hepatocellular apoptosis and innate immune system-mediated inflammation. These responses are orchestrated by signaling pathways that can be activated by lipids, directly or indirectly. In this review, we discuss palmitate- and lysophosphatidylcholine (LPC)-induced upregulation of p53-upregulated modulator of apoptosis and cell-surface expression of the death receptor TNF-related apoptosis-inducing ligand receptor 2. Next, we review the activation of stress-induced kinases, mixed lineage kinase 3, and c-Jun N-terminal kinase, and the activation of endoplasmic reticulum stress response and its downstream proapoptotic effector, CAAT/enhancer binding homologous protein, by palmitate and LPC. Moreover, the activation of these stress signaling pathways is linked to the release of proinflammatory, proangiogenic, and profibrotic extracellular vesicles by stressed hepatocytes. This review discusses the signaling pathways induced by lethal and sublethal lipid overload that contribute to the pathogenesis of NAFLD.
Keywords: apoptosis; cell death; cell signaling; exosomes; extracellular vesicles; fatty acids; lipotoxicity; lysophosphatidylcholine; nonalcoholic fatty liver disease; nonalcoholic steatohepatitis; steatohepatitis.
Copyright © 2016 by the American Society for Biochemistry and Molecular Biology, Inc.
Figures
References
-
- Unger R. H., Clark G. O., Scherer P. E., and Orci L.. 2010. Lipid homeostasis, lipotoxicity and the metabolic syndrome. Biochim. Biophys. Acta. 1801: 209–214. - PubMed
-
- Rinella M. E. 2015. Nonalcoholic fatty liver disease: a systematic review. JAMA. 313: 2263–2273. - PubMed
-
- Puri P., Baillie R. A., Wiest M. M., Mirshahi F., Choudhury J., Cheung O., Sargeant C., Contos M. J., and Sanyal A. J.. 2007. A lipidomic analysis of nonalcoholic fatty liver disease. Hepatology. 46: 1081–1090. - PubMed
-
- Gorden D. L., Ivanova P. T., Myers D. S., McIntyre J. O., VanSaun M. N., Wright J. K., Matrisian L. M., and Brown H. A.. 2011. Increased diacylglycerols characterize hepatic lipid changes in progression of human nonalcoholic fatty liver disease; comparison to a murine model. PLoS One. 6: e22775. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
