

Genetic and environmental influences interact with age and sex in shaping the human methylome
- PMID: 27051996
- PMCID: PMC4820961
- DOI: 10.1038/ncomms11115
Genetic and environmental influences interact with age and sex in shaping the human methylome
Abstract
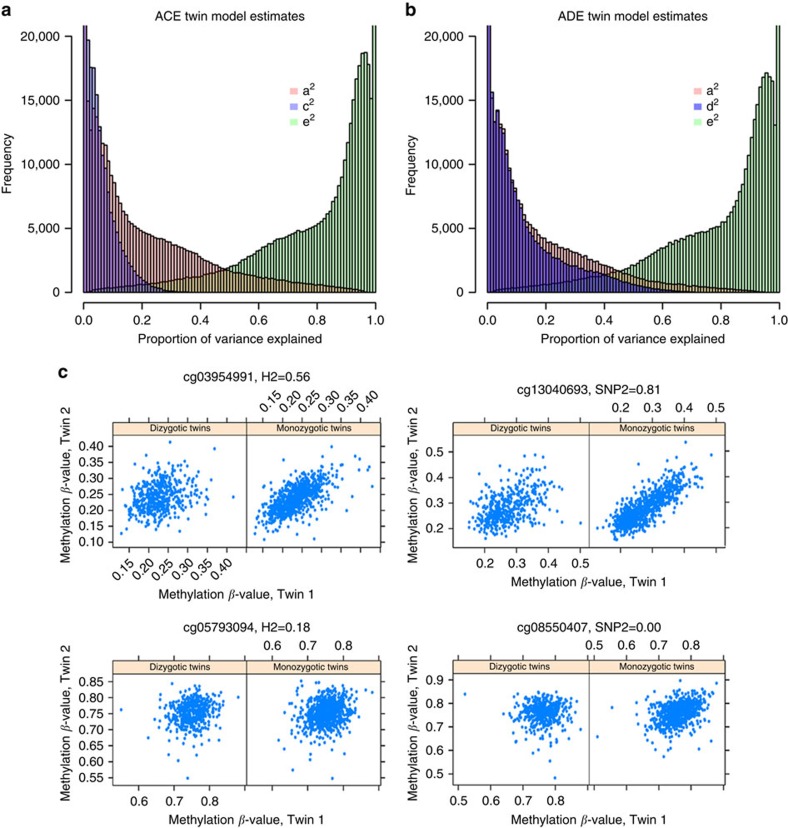
The methylome is subject to genetic and environmental effects. Their impact may depend on sex and age, resulting in sex- and age-related physiological variation and disease susceptibility. Here we estimate the total heritability of DNA methylation levels in whole blood and estimate the variance explained by common single nucleotide polymorphisms at 411,169 sites in 2,603 individuals from twin families, to establish a catalogue of between-individual variation in DNA methylation. Heritability estimates vary across the genome (mean=19%) and interaction analyses reveal thousands of sites with sex-specific heritability as well as sites where the environmental variance increases with age. Integration with previously published data illustrates the impact of genome and environment across the lifespan at methylation sites associated with metabolic traits, smoking and ageing. These findings demonstrate that our catalogue holds valuable information on locations in the genome where methylation variation between people may reflect disease-relevant environmental exposures or genetic variation.
Figures






References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
