

A mouse model of a human congenital disorder of glycosylation caused by loss of PMM2
- PMID: 27053713
- PMCID: PMC5081049
- DOI: 10.1093/hmg/ddw085
A mouse model of a human congenital disorder of glycosylation caused by loss of PMM2
Abstract
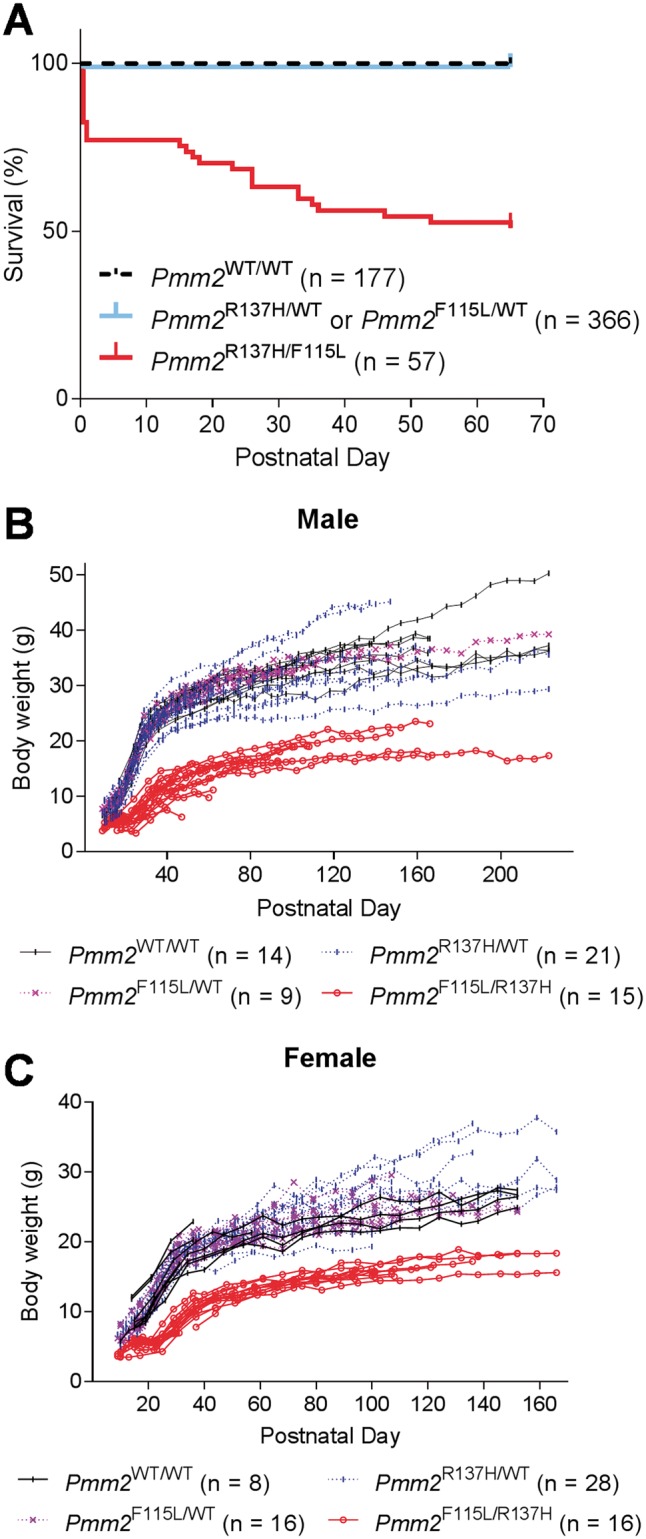
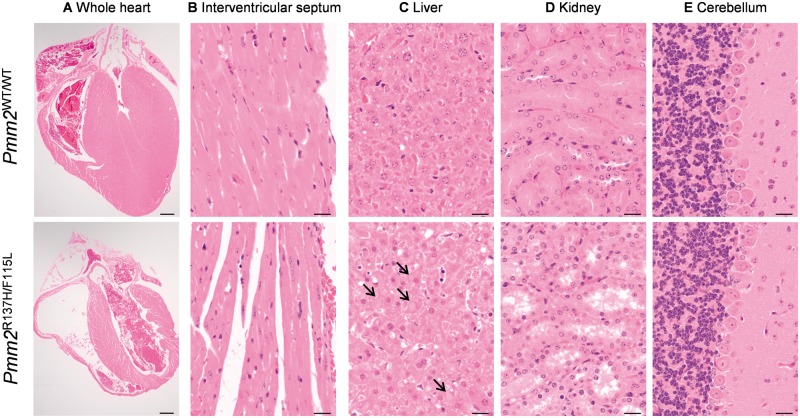
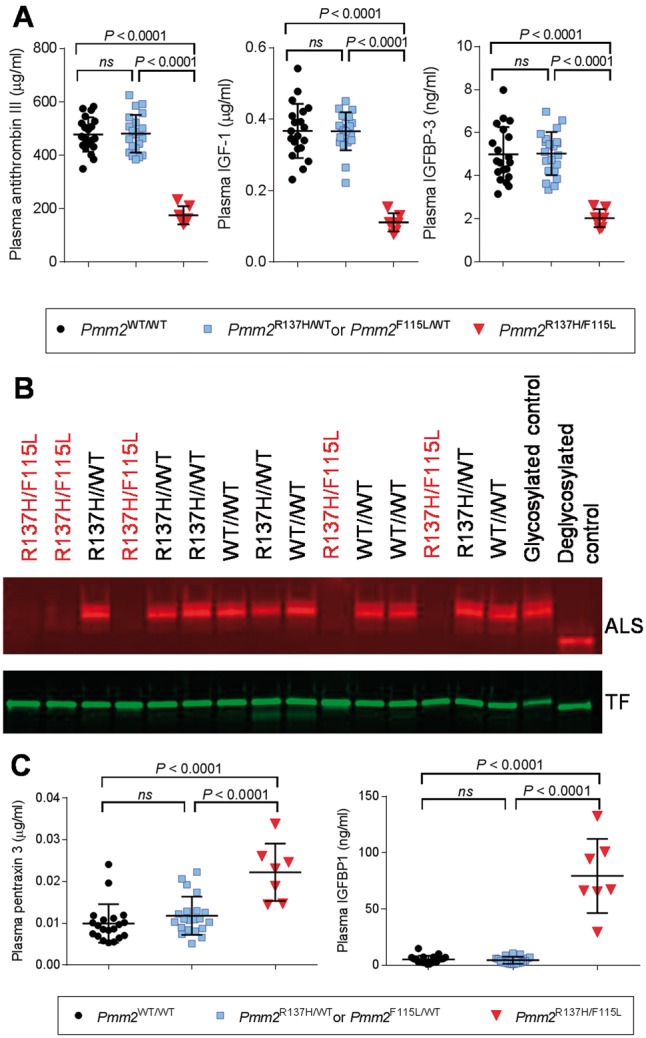
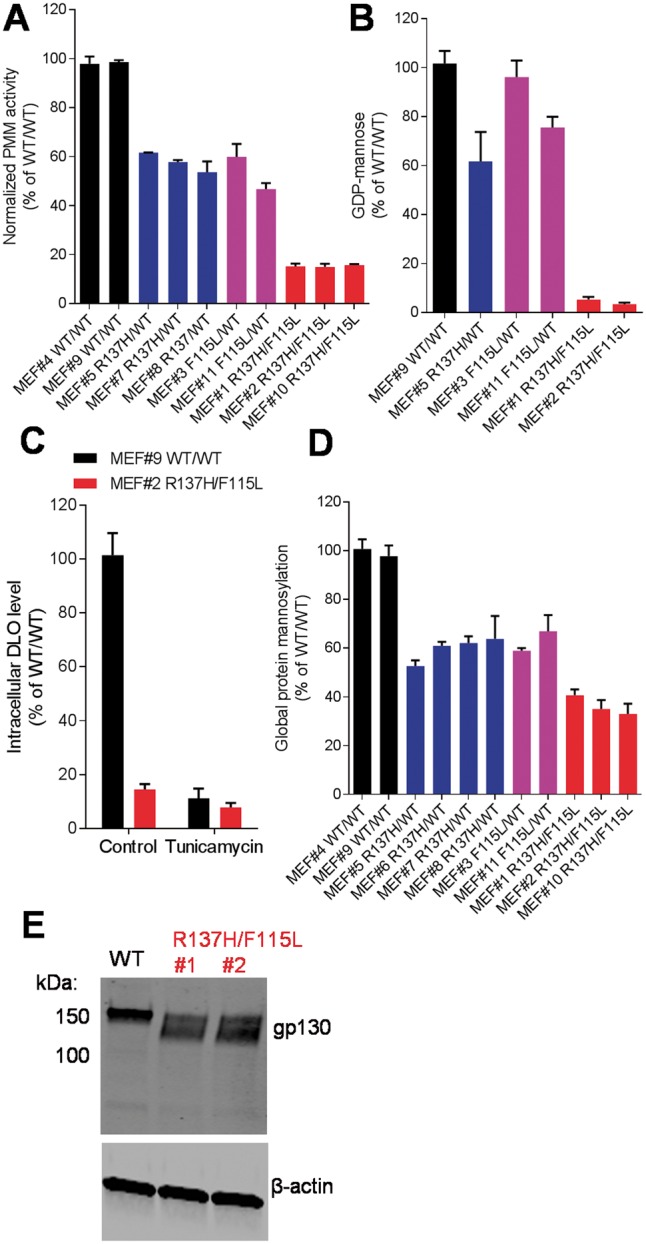
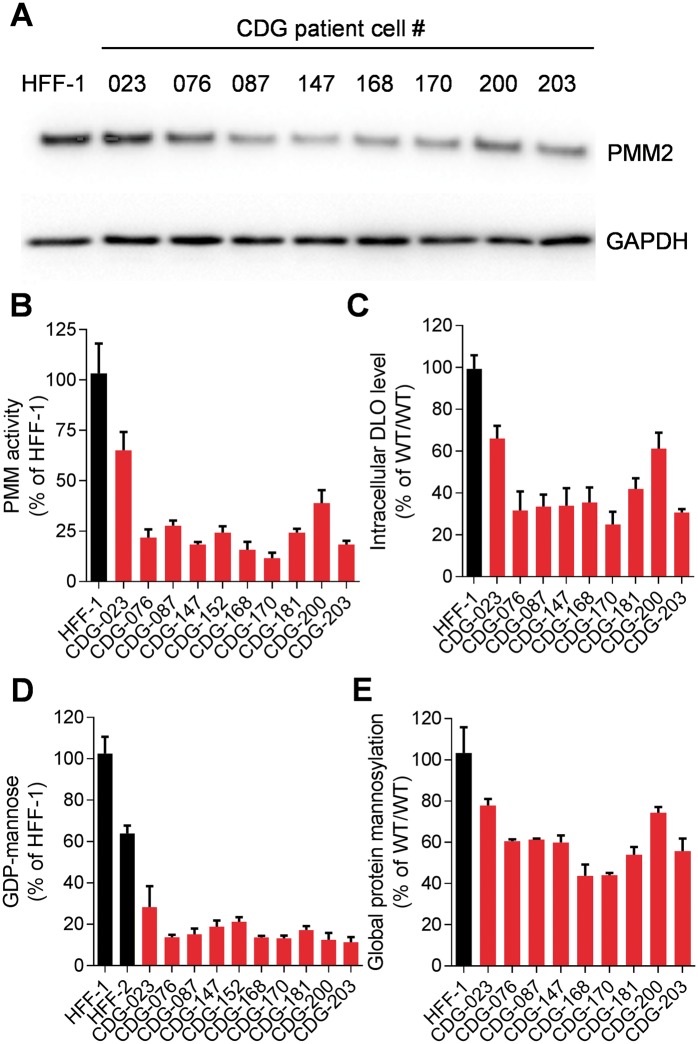
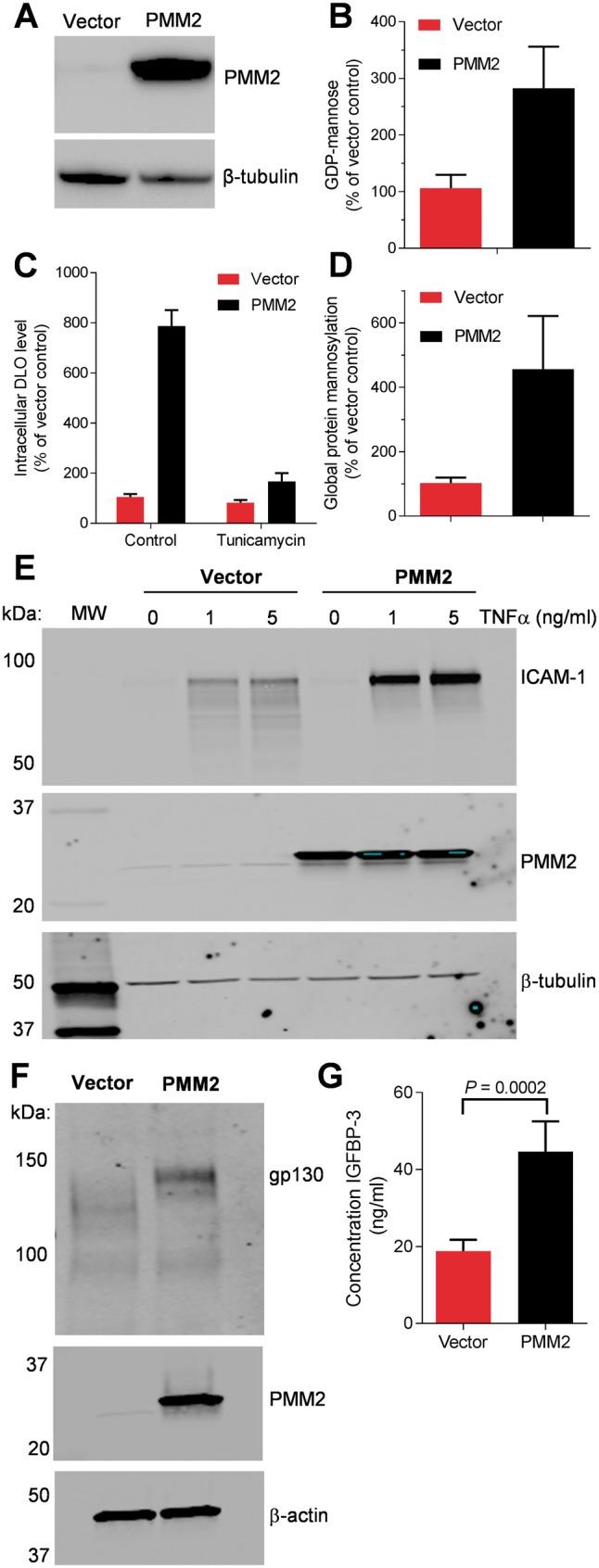
The most common congenital disorder of glycosylation (CDG), phosphomannomutase 2 (PMM2)-CDG, is caused by mutations in PMM2 that limit availability of mannose precursors required for protein N-glycosylation. The disorder has no therapy and there are no models to test new treatments. We generated compound heterozygous mice with the R137H and F115L mutations in Pmm2 that correspond to the most prevalent alleles found in patients with PMM2-CDG. Many Pmm2R137H/F115L mice died prenatally, while survivors had significantly stunted growth. These animals and cells derived from them showed protein glycosylation deficiencies similar to those found in patients with PMM2-CDG. Growth-related glycoproteins insulin-like growth factor (IGF) 1, IGF binding protein-3 and acid-labile subunit, along with antithrombin III, were all deficient in Pmm2R137H/F115L mice, but their levels in heterozygous mice were comparable to wild-type (WT) littermates. These imbalances, resulting from defective glycosylation, are likely the cause of the stunted growth seen both in our model and in PMM2-CDG patients. Both Pmm2R137H/F115L mouse and PMM2-CDG patient-derived fibroblasts displayed reductions in PMM activity, guanosine diphosphate mannose, lipid-linked oligosaccharide precursor and total cellular protein glycosylation, along with hypoglycosylation of a new endogenous biomarker, glycoprotein 130 (gp130). Over-expression of WT-PMM2 in patient-derived fibroblasts rescued all these defects, showing that restoration of mutant PMM2 activity is a viable therapeutic strategy. This functional mouse model of PMM2-CDG, in vitro assays and identification of the novel gp130 biomarker all shed light on the human disease, and moreover, provide the essential tools to test potential therapeutics for this untreatable disease.
© The Author 2016. Published by Oxford University Press.
Figures
References
-
- Freeze H.H, Schachter H. (2009) Genetic disorders of glycosylation In Varki A., Cummings R.D., Esko J.D., Freeze H.H., Stanley P., Bertozzi C.R., Hart G.W., Etzler M.E. (eds), Essentials of Glycobiology. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, Chapter 42. - PubMed
-
- Cylwi B., Naklicki M., Chrostek L., Gruszewska E. (2013) Congenital disorders of glycosylation. Part I. Defects of protein N-glycosylation. Acta Biochim. Pol, 20, 151–161. - PubMed
-
- Jaeken J., van Eijk H.G., van der Heul C., Corbeel L., Eeckels R., Eggermont E. (1984) Sialic acid-deficient serum and cerebrospinal fluid transferrin in a newly recognized genetic syndrome. Clin. Chim. Acta, 144, 245–247. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
