

Cancer drug resistance: redox resetting renders a way
- PMID: 27057637
- PMCID: PMC5173169
- DOI: 10.18632/oncotarget.8600
Cancer drug resistance: redox resetting renders a way
Abstract
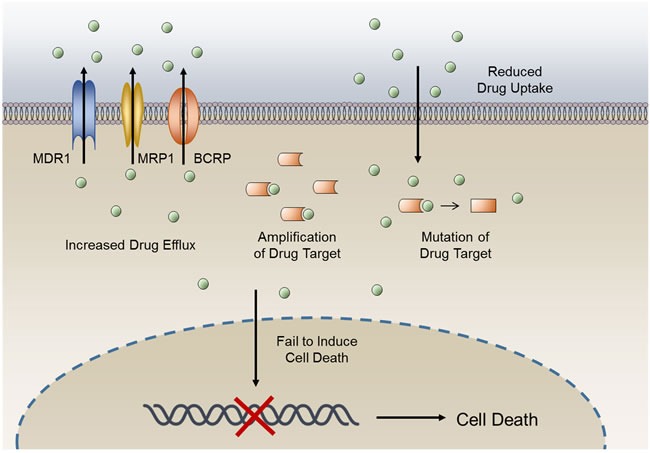
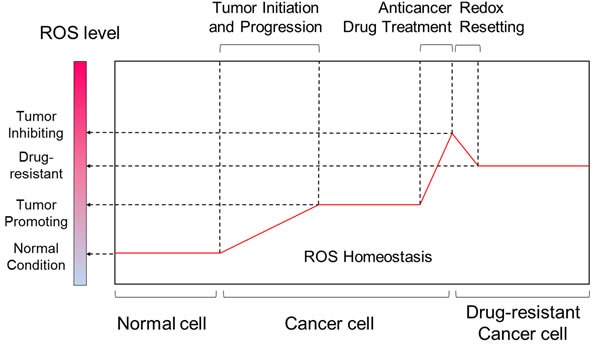
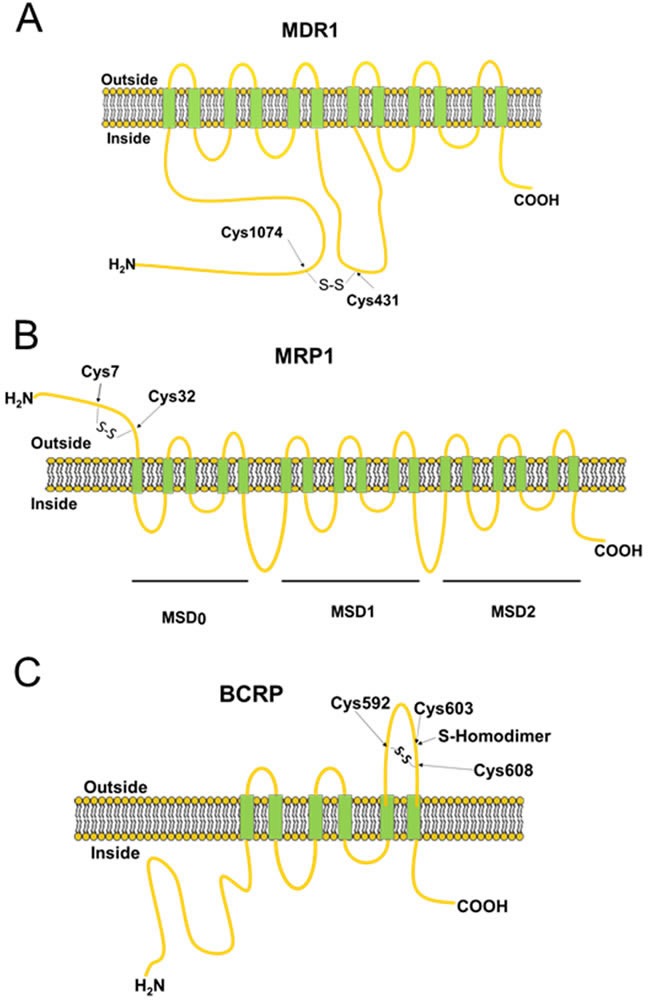
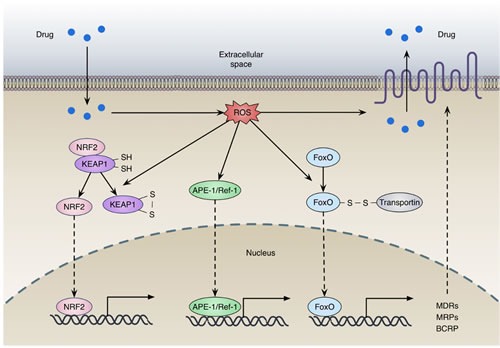
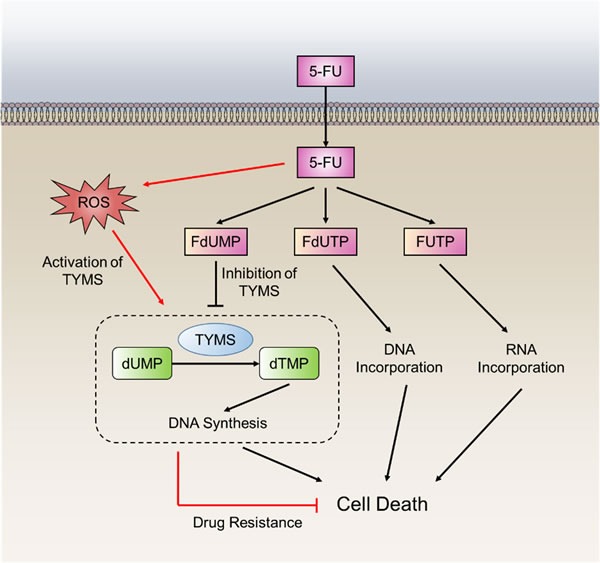
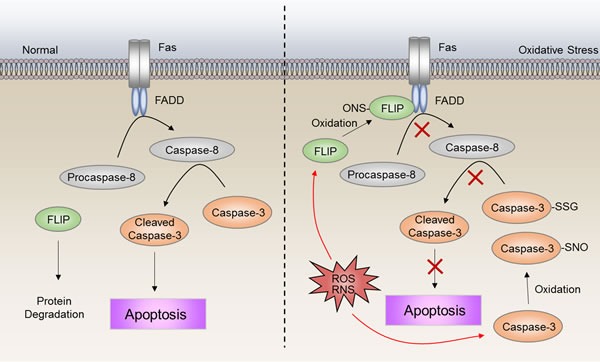
Disruption of redox homeostasis is a crucial factor in the development of drug resistance, which is a major problem facing current cancer treatment. Compared with normal cells, tumor cells generally exhibit higher levels of reactive oxygen species (ROS), which can promote tumor progression and development. Upon drug treatment, some tumor cells can undergo a process of 'Redox Resetting' to acquire a new redox balance with higher levels of ROS accumulation and stronger antioxidant systems. Evidence has accumulated showing that the 'Redox Resetting' enables cancer cells to become resistant to anticancer drugs by multiple mechanisms, including increased rates of drug efflux, altered drug metabolism and drug targets, activated prosurvival pathways and inefficient induction of cell death. In this article, we provide insight into the role of 'Redox Resetting' on the emergence of drug resistance that may contribute to pharmacological modulation of resistance.
Keywords: cancer therapy; drug efflux; drug resistance; oxidative stress; redox modifications.
Conflict of interest statement
There is no any competing interest.
Figures
References
-
- Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer drug resistance: an evolving paradigm. Nature reviews Cancer. 2013;13:714–726. - PubMed
-
- Bock C, Lengauer T. Managing drug resistance in cancer: lessons from HIV therapy. Nature reviews Cancer. 2012;12:494–501. - PubMed
-
- Tanaka H, Matsumura I, Ezoe S, Satoh Y, Sakamaki T, Albanese C, Machii T, Pestell RG, Kanakura Y. E2F1 and c-Myc potentiate apoptosis through inhibition of NF-kappaB activity that facilitates MnSOD-mediated ROS elimination. Molecular cell. 2002;9:1017–1029. - PubMed
-
- Gorrini C, Harris IS, Mak TW. Modulation of oxidative stress as an anticancer strategy. Nature reviews Drug discovery. 2013;12:931–947. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
