

Hepcidin: A Promising Therapeutic Target for Iron Disorders: A Systematic Review
- PMID: 27057839
- PMCID: PMC4998755
- DOI: 10.1097/MD.0000000000003150
Hepcidin: A Promising Therapeutic Target for Iron Disorders: A Systematic Review
Abstract
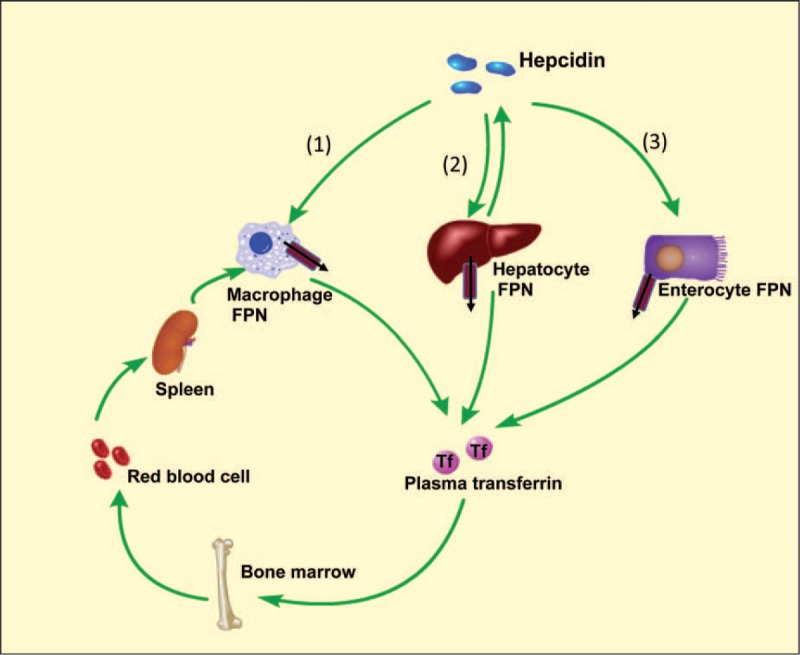
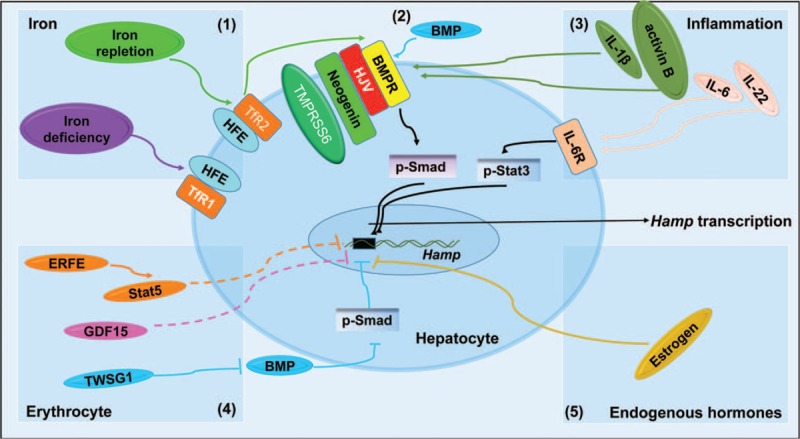
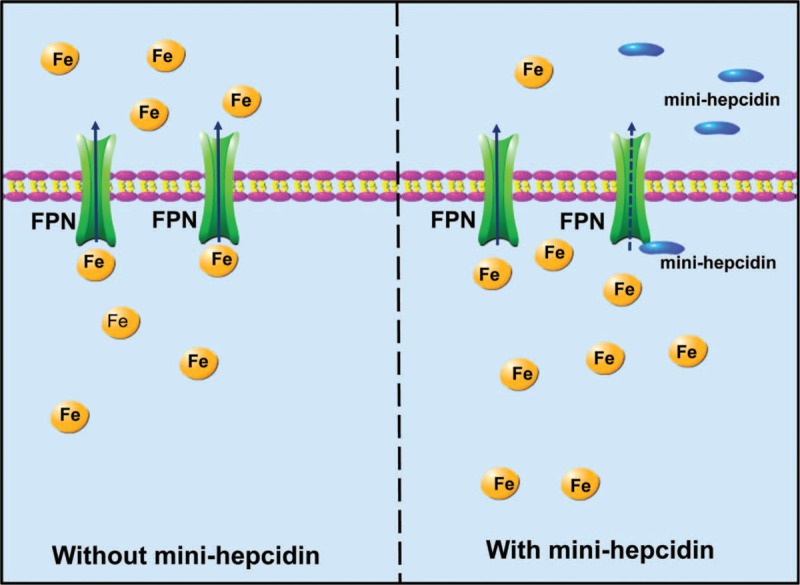
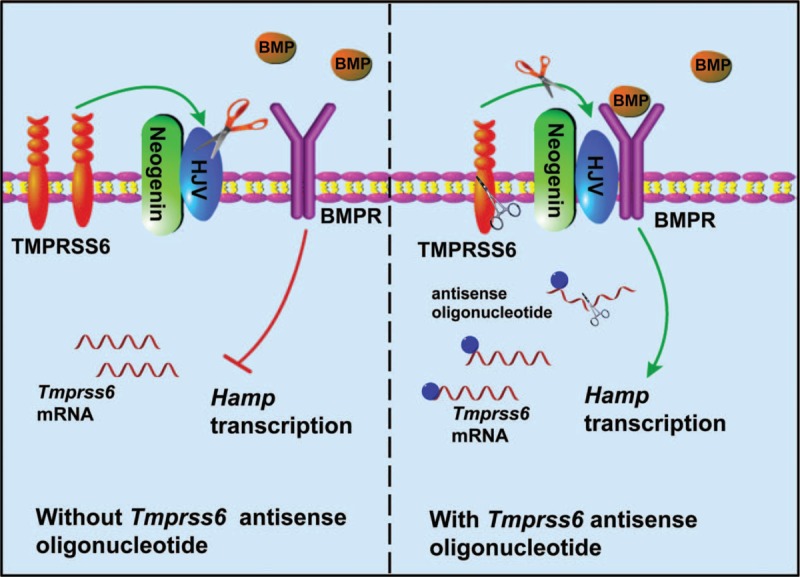
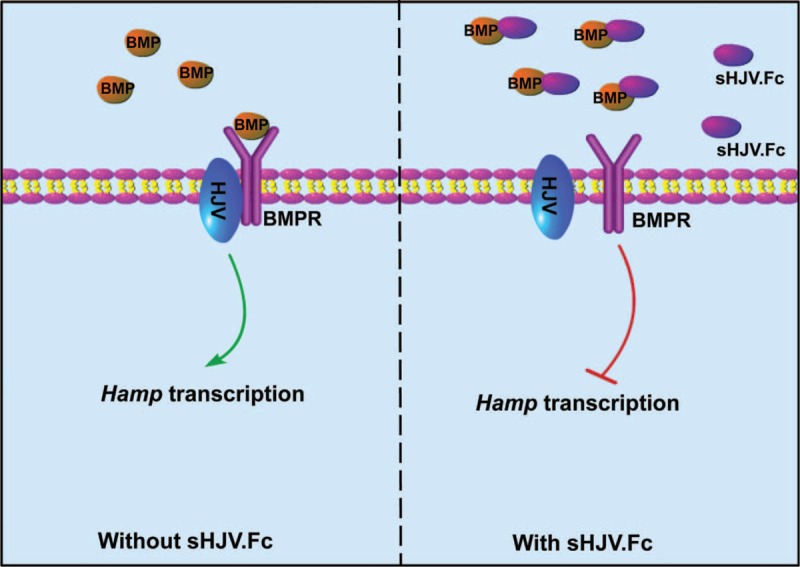
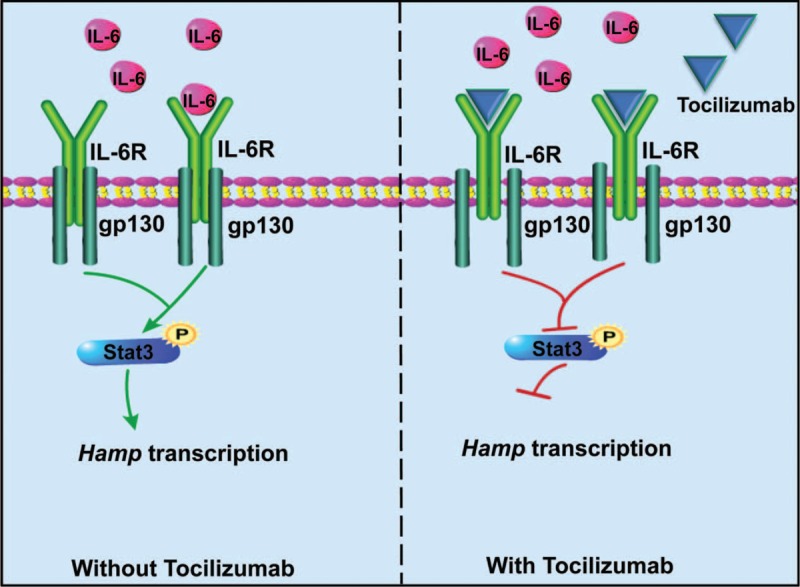
Iron is required for most forms of organisms, and it is the most essential element for the functions of many iron-containing proteins involved in oxygen transport, cellular respiration, DNA replication, and so on. Disorders of iron metabolism are associated with diverse diseases, including anemias (e.g., iron-deficiency anemia and anemia of chronic diseases) and iron overload diseases, such as hereditary hemochromatosis and β-thalassemia. Hepcidin (encoded by Hamp gene) is a peptide hormone synthesized by hepatocytes, and it plays an important role in regulating the systematic iron homeostasis. As the systemic iron regulator, hepcidin, not only controls dietary iron absorption and iron egress out of iron storage cells, but also induces iron redistribution in various organs. Deregulated hepcidin is often seen in a variety of iron-related diseases including anemias and iron overload disorders. In the case of iron overload disorders (e.g., hereditary hemochromatosis and β-thalassemia), hepatic hepcidin concentration is significantly reduced.Since hepcidin deregulation is responsible for iron disorder-associated diseases, the purpose of this review is to summarize the recent findings on therapeutics targeting hepcidin.Continuous efforts have been made to search for hepcidin mimics and chemical compounds that could be used to increase hepcidin level. Here, a literature search was conducted in PubMed, and research papers relevant to hepcidin regulation or hepcidin-centered therapeutic work were reviewed. On the basis of literature search, we recapitulated recent findings on therapeutic studies targeting hepcidin, including agonists and antagonists to modulate hepcidin expression or its downstream signaling. We also discussed the molecular mechanisms by which hepcidin level and iron metabolism are modulated.Elevating hepcidin concentration is an optimal strategy to ameliorate iron overload diseases, and also to relieve β-thalassemia phenotypes by improving ineffective erythropoiesis. Relative to the current conventional therapies, such as phlebotomy and blood transfusion, therapeutics targeting hepcidin would open a new avenue for treatment of iron-related diseases.
Conflict of interest statement
The authors declare no competing financial interests. The authors have nothing to disclose.
Figures
References
-
- Evstatiev R, Gasche C. Iron sensing and signalling. Gut 2012; 61:933–952. - PubMed
-
- Raiten DJ. Iron: current landscape and efforts to address a complex issue in a complex world. J Pediatr 2015; 167:S3–7. - PubMed
-
- Porter JB. Optimizing iron chelation strategies in beta-thalassaemia major. Blood Rev 2009; 23:3–S7. - PubMed
-
- Pippard MJ, Warner GT, Callender ST, et al. Iron-absorption and loading in beta-thalassemia intermedia. Lancet 1979; 2:819–821. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
