

Polymorphic tandem repeats within gene promoters act as modifiers of gene expression and DNA methylation in humans
- PMID: 27060133
- PMCID: PMC4857002
- DOI: 10.1093/nar/gkw219
Polymorphic tandem repeats within gene promoters act as modifiers of gene expression and DNA methylation in humans
Abstract
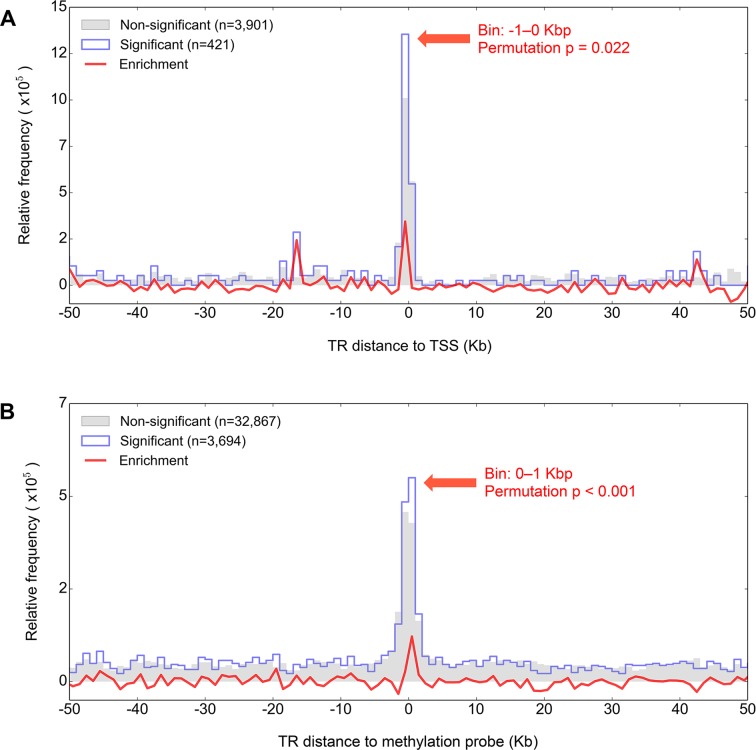
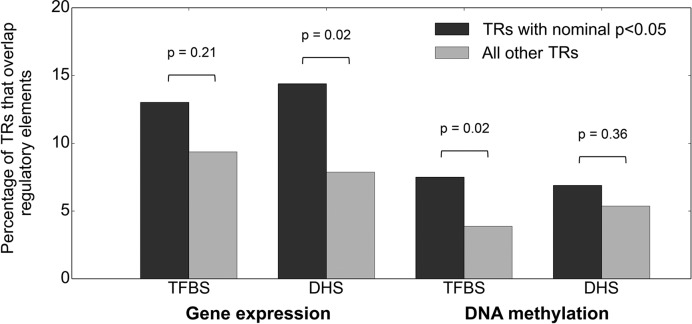
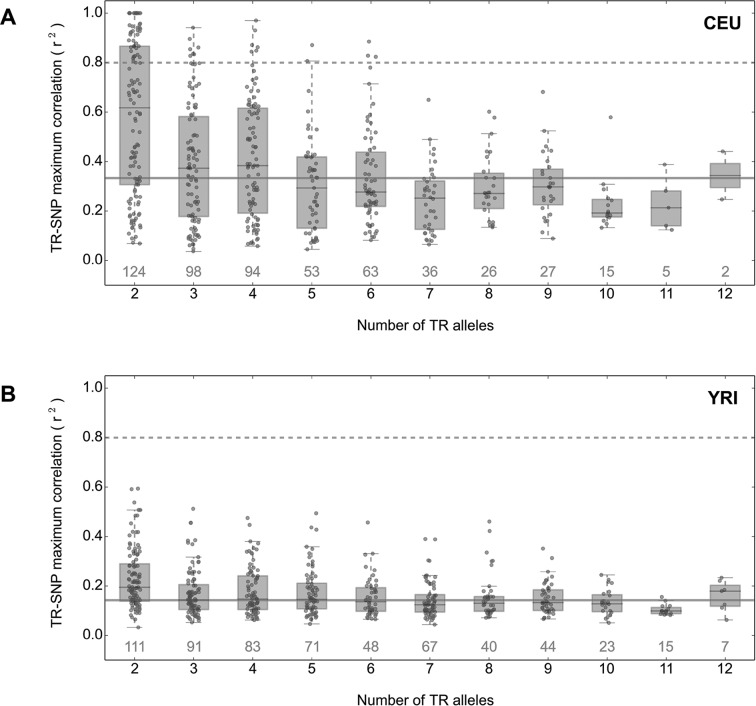
Despite representing an important source of genetic variation, tandem repeats (TRs) remain poorly studied due to technical difficulties. We hypothesized that TRs can operate as expression (eQTLs) and methylation (mQTLs) quantitative trait loci. To test this we analyzed the effect of variation at 4849 promoter-associated TRs, genotyped in 120 individuals, on neighboring gene expression and DNA methylation. Polymorphic promoter TRs were associated with increased variance in local gene expression and DNA methylation, suggesting functional consequences related to TR variation. We identified >100 TRs associated with expression/methylation levels of adjacent genes. These potential eQTL/mQTL TRs were enriched for overlaps with transcription factor binding and DNaseI hypersensitivity sites, providing a rationale for their effects. Moreover, we showed that most TR variants are poorly tagged by nearby single nucleotide polymorphisms (SNPs) markers, indicating that many functional TR variants are not effectively assayed by SNP-based approaches. Our study assigns biological significance to TR variations in the human genome, and suggests that a significant fraction of TR variations exert functional effects via alterations of local gene expression or epigenetics. We conclude that targeted studies that focus on genotyping TR variants are required to fully ascertain functional variation in the genome.
© The Author(s) 2016. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures
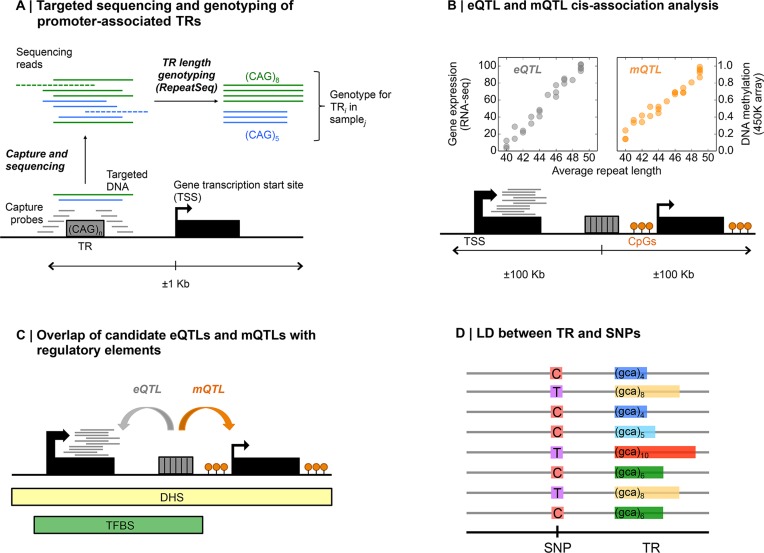
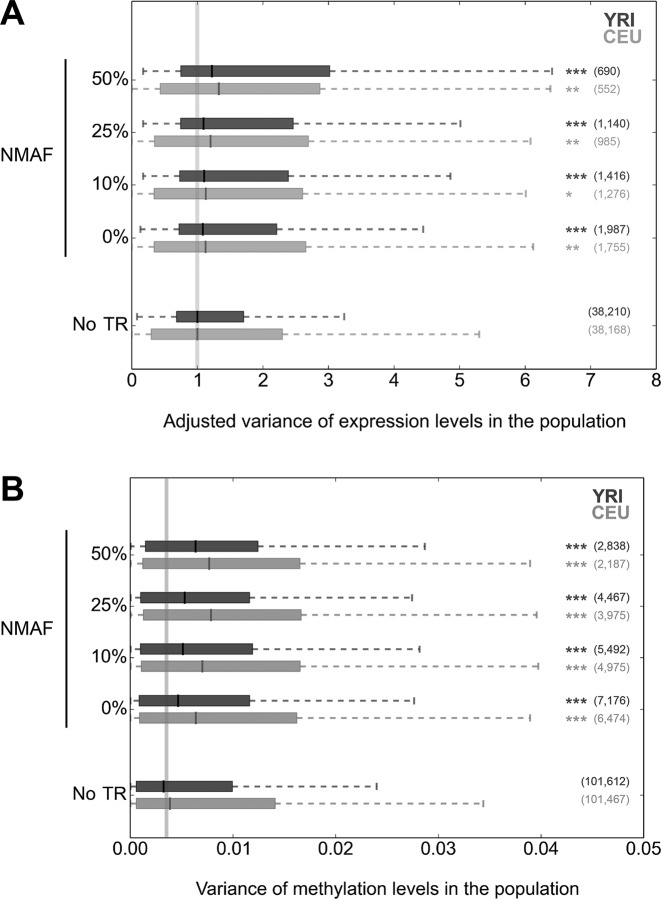
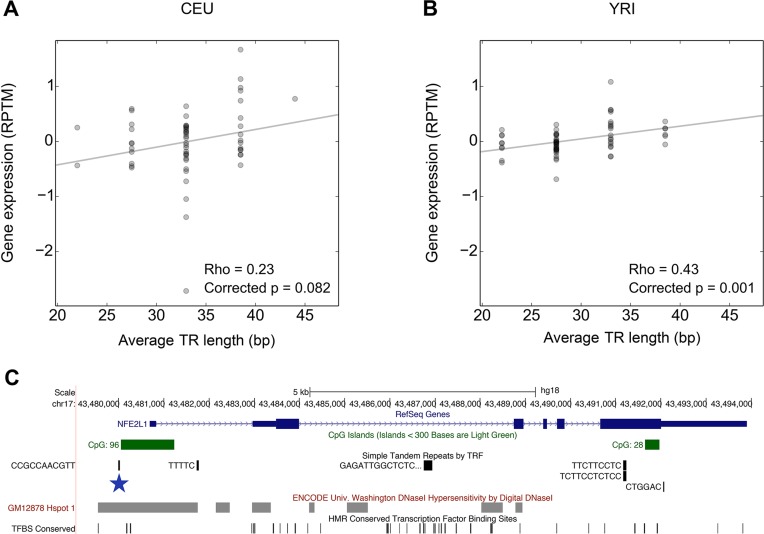
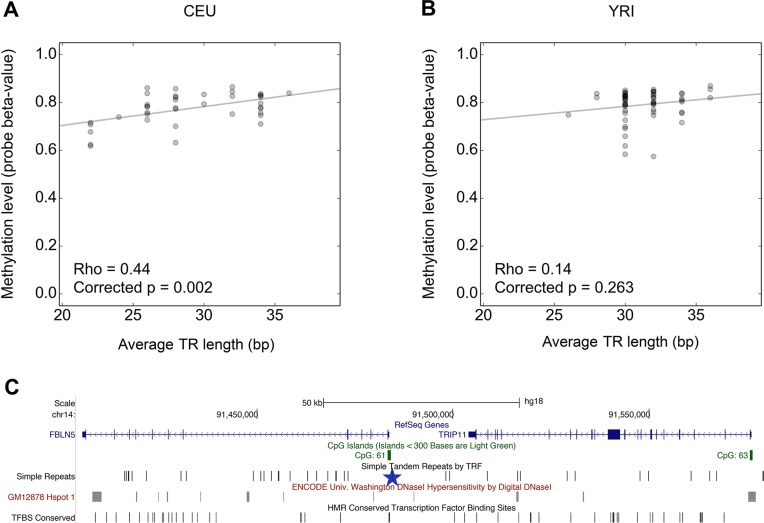
References
-
- Lander E.S., Linton L.M., Birren B., Nusbaum C., Zody M.C., Baldwin J., Devon K., Dewar K., Doyle M., FitzHugh W., et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. - PubMed
-
- Kondrashov A.S. Direct estimates of human per nucleotide mutation rates at 20 loci causing Mendelian diseases. Hum. Mutat. 2003;21:12–27. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
