

Control of plasma membrane lipid homeostasis by the extended synaptotagmins
- PMID: 27065097
- PMCID: PMC4848133
- DOI: 10.1038/ncb3339
Control of plasma membrane lipid homeostasis by the extended synaptotagmins
Abstract
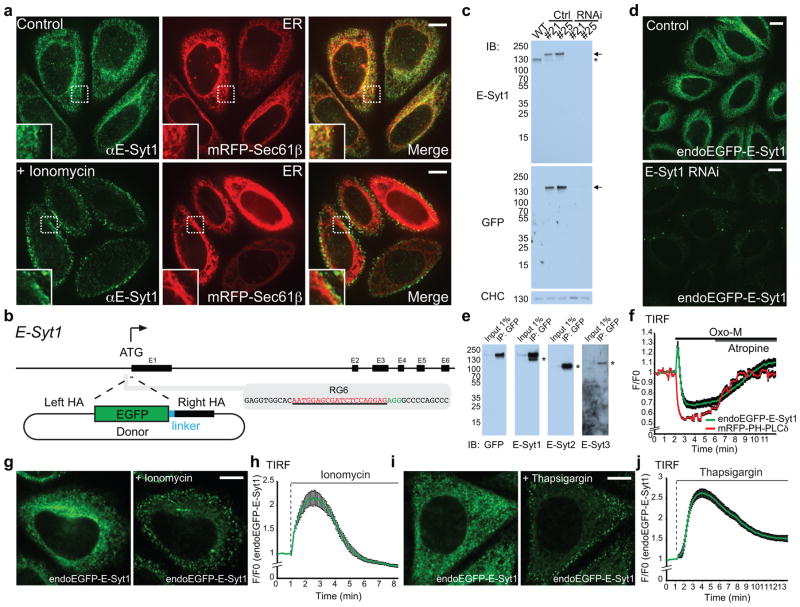
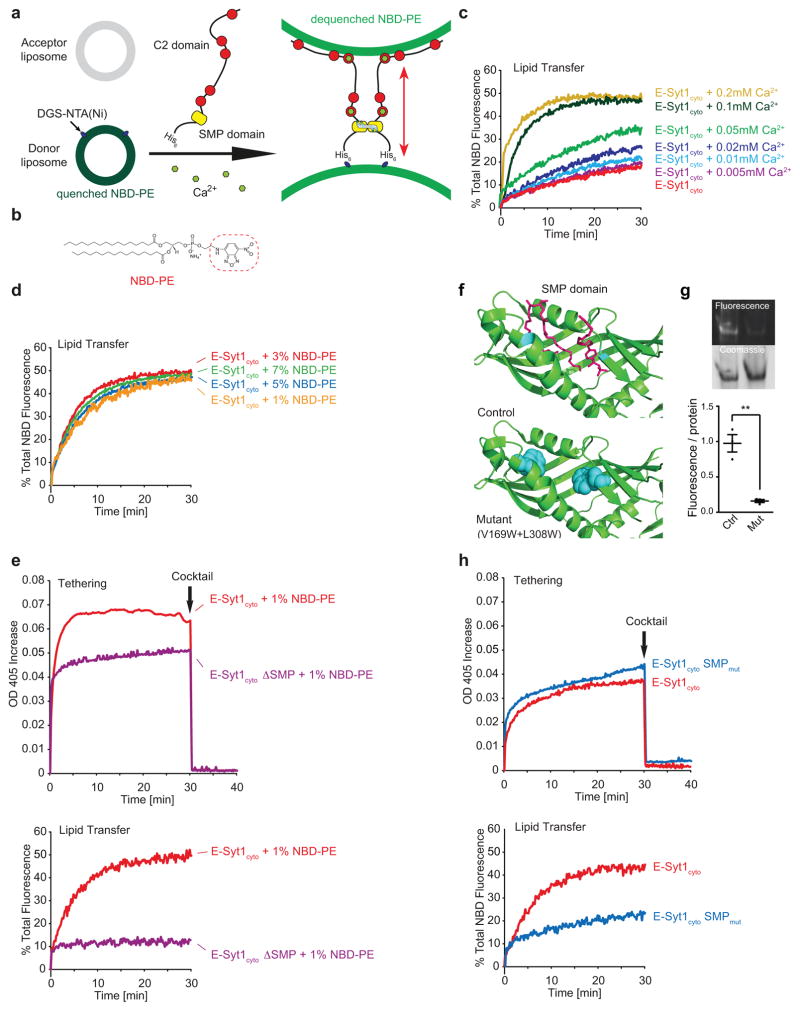
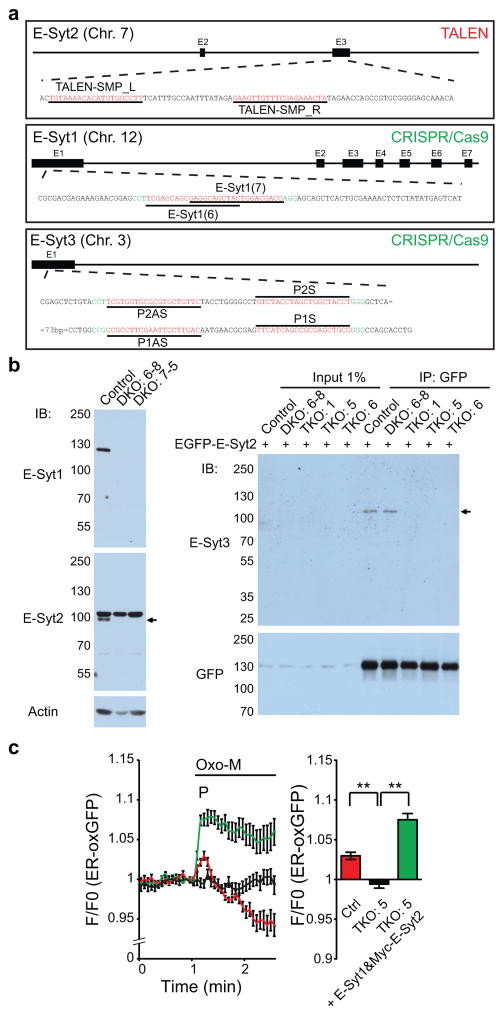
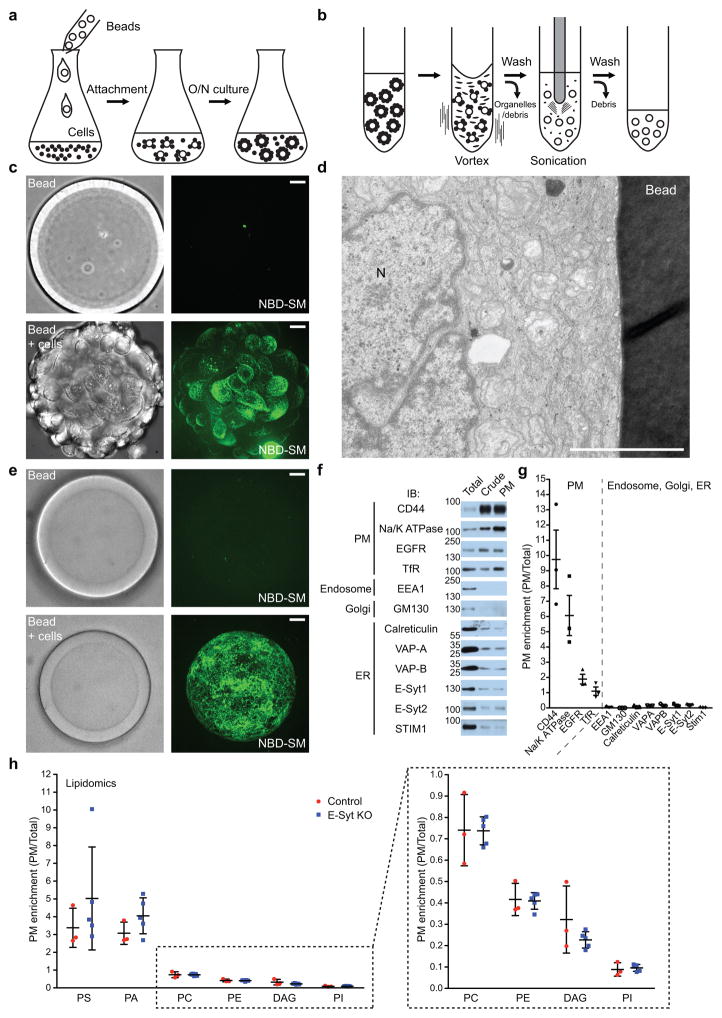
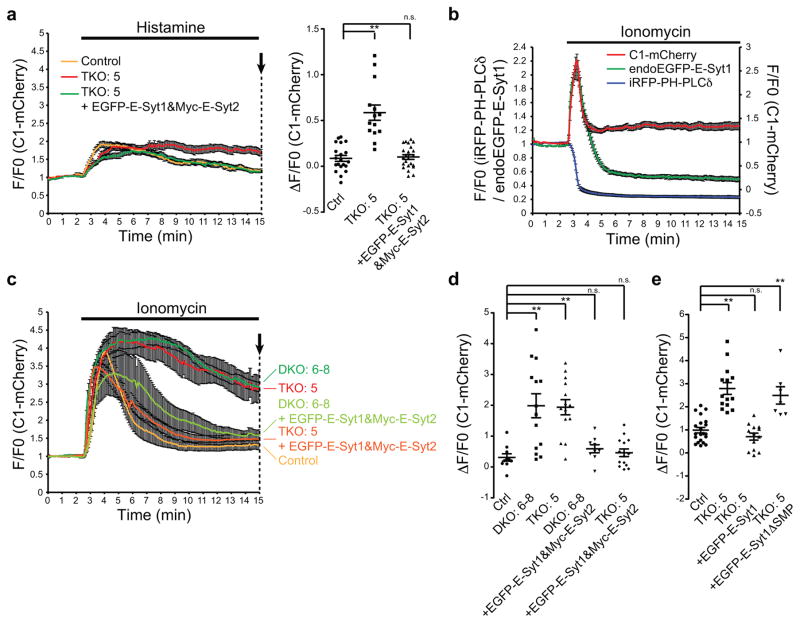
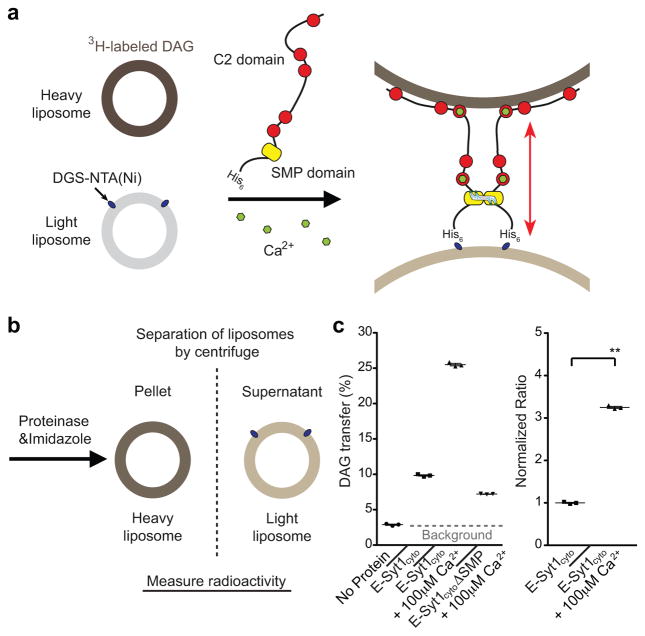
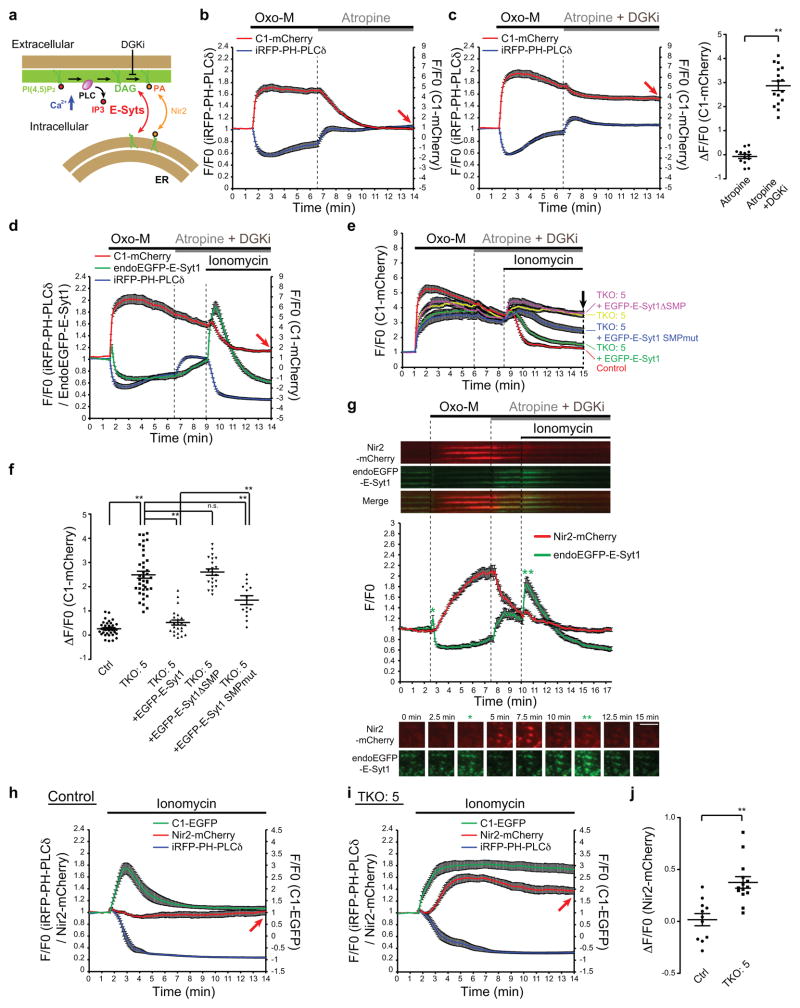
Acute metabolic changes in plasma membrane (PM) lipids, such as those mediating signalling reactions, are rapidly compensated by homeostatic responses whose molecular basis is poorly understood. Here we show that the extended synaptotagmins (E-Syts), endoplasmic reticulum (ER) proteins that function as PtdIns(4,5)P2- and Ca(2+)-regulated tethers to the PM, participate in these responses. E-Syts transfer glycerolipids between bilayers in vitro, and this transfer requires Ca(2+) and their lipid-harbouring SMP domain. Genome-edited cells lacking E-Syts do not exhibit abnormalities in the major glycerolipids at rest, but exhibit enhanced and sustained accumulation of PM diacylglycerol following PtdIns(4,5)P2 hydrolysis by PLC activation, which can be rescued by expression of E-Syt1, but not by mutant E-Syt1 lacking the SMP domain. The formation of E-Syt-dependent ER-PM tethers in response to stimuli that cleave PtdIns(4,5)P2 and elevate Ca(2+) may help reverse accumulation of diacylglycerol in the PM by transferring it to the ER for metabolic recycling.
Figures
Comment in
-
Directing lipid transport at membrane contact sites.Nat Cell Biol. 2016 Apr 27;18(5):461-3. doi: 10.1038/ncb3345. Nat Cell Biol. 2016. PMID: 27117329
References
-
- Holthuis JC, Levine TP. Lipid traffic: floppy drives and a superhighway. Nature reviews. Molecular cell biology. 2005;6:209–220. - PubMed
-
- Elbaz Y, Schuldiner M. Staying in touch: the molecular era of organelle contact sites. Trends in biochemical sciences. 2011;36:616–623. - PubMed
-
- Stefan CJ, et al. Osh proteins regulate phosphoinositide metabolism at ER-plasma membrane contact sites. Cell. 2011;144:389–401. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
