

Epilepsy with auditory features: A heterogeneous clinico-molecular disease
- PMID: 27066544
- PMCID: PMC4821078
- DOI: 10.1212/NXG.0000000000000005
Epilepsy with auditory features: A heterogeneous clinico-molecular disease
Abstract
Objective: To identify novel genes implicated in epilepsy with auditory features (EAF) in phenotypically heterogeneous families with unknown molecular basis.
Methods: We identified 15 probands with EAF in whom an LGI1 mutation had been excluded. We performed electroclinical phenotyping on all probands and available affected relatives. We used whole-exome sequencing (WES) in 20 individuals with EAF (including all the probands and 5 relatives) to identify single nucleotide variants, small insertions/deletions, and copy number variants.
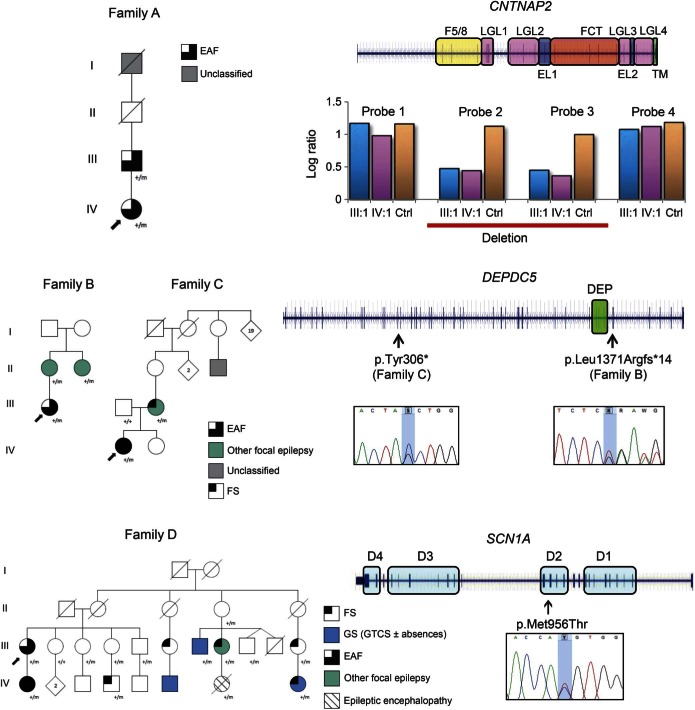
Results: WES revealed likely pathogenic variants in genes that had not been previously associated with EAF: a CNTNAP2 intragenic deletion, 2 truncating mutations of DEPDC5, and a missense SCN1A change.
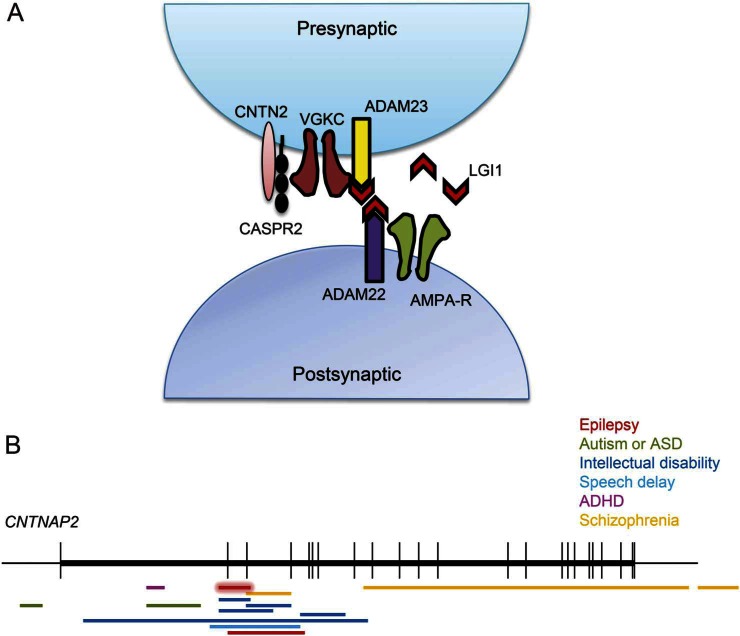
Conclusions: EAF is a clinically and molecularly heterogeneous disease. The association of EAF with CNTNAP2, DEPDC5, and SCN1A mutations widens the phenotypic spectrum related to these genes. CNTNAP2 encodes CASPR2, a member of the voltage-gated potassium channel complex in which LGI1 plays a role. The finding of a CNTNAP2 deletion emphasizes the importance of this complex in EAF and shows biological convergence.
Figures
Similar articles
-
Epilepsy with auditory features: Contribution of known genes in 112 patients.Seizure. 2021 Feb;85:115-118. doi: 10.1016/j.seizure.2020.12.015. Epub 2020 Dec 31. Seizure. 2021. PMID: 33453592
-
SCN1A mutations in focal epilepsy with auditory features: widening the spectrum of GEFS plus.Epileptic Disord. 2019 Apr 1;21(2):185-191. doi: 10.1684/epd.2019.1046. Epileptic Disord. 2019. PMID: 30977726
-
CNTNAP2 mutations and autosomal dominant epilepsy with auditory features.Epilepsy Res. 2018 Jan;139:51-53. doi: 10.1016/j.eplepsyres.2017.11.006. Epub 2017 Nov 21. Epilepsy Res. 2018. PMID: 29179159
-
Genetic and epigenetic mechanisms of epilepsy: a review.Neuropsychiatr Dis Treat. 2017 Jul 13;13:1841-1859. doi: 10.2147/NDT.S142032. eCollection 2017. Neuropsychiatr Dis Treat. 2017. PMID: 28761347 Free PMC article. Review.
-
Connecting the CNTNAP2 Networks with Neurodevelopmental Disorders.Mol Syndromol. 2015 Feb;6(1):7-22. doi: 10.1159/000371594. Epub 2015 Feb 3. Mol Syndromol. 2015. PMID: 25852443 Free PMC article. Review.
Cited by
-
Spotlight on the June 2015 issue.Neurol Genet. 2015 Jul 2;1(1):e11. doi: 10.1212/NXG.0000000000000011. eCollection 2015 Jun. Neurol Genet. 2015. PMID: 27066540 Free PMC article.
-
Enhanced copy number variants detection from whole-exome sequencing data using EXCAVATOR2.Nucleic Acids Res. 2016 Nov 16;44(20):e154. doi: 10.1093/nar/gkw695. Epub 2016 Aug 9. Nucleic Acids Res. 2016. PMID: 27507884 Free PMC article.
-
What is the impact of a novel DEPDC5 variant on an infant with focal epilepsy: a case report.BMC Pediatr. 2022 Jul 30;22(1):459. doi: 10.1186/s12887-022-03515-8. BMC Pediatr. 2022. PMID: 35907814 Free PMC article.
-
Loss of function mutations in CCDC32 cause a congenital syndrome characterized by craniofacial, cardiac and neurodevelopmental anomalies.Hum Mol Genet. 2020 Jun 3;29(9):1489-1497. doi: 10.1093/hmg/ddaa073. Hum Mol Genet. 2020. PMID: 32307552 Free PMC article.
-
DEPDC5 Variants Associated Malformations of Cortical Development and Focal Epilepsy With Febrile Seizure Plus/Febrile Seizures: The Role of Molecular Sub-Regional Effect.Front Neurosci. 2020 Aug 11;14:821. doi: 10.3389/fnins.2020.00821. eCollection 2020. Front Neurosci. 2020. PMID: 32848577 Free PMC article.
References
-
- Berg AT, Berkovic SF, Brodie MJ, et al. Revised terminology and concepts for organization of seizures and epilepsies: report of the ILAE Commission on Classification and Terminology, 2005–2009. Epilepsia 2010;51:676–685. - PubMed
-
- Morante-Redolat JM, Gorostidi-Pagola A, Piquer-Sirerol S, et al. Mutations in the LGI1/Epitempin gene on 10q24 cause autosomal dominant lateral temporal epilepsy. Hum Mol Genet 2002;11:1119–1128. - PubMed
LinkOut - more resources
Full Text Sources
Medical