

Expanding genotype/phenotype of neuromuscular diseases by comprehensive target capture/NGS
- PMID: 27066551
- PMCID: PMC4807910
- DOI: 10.1212/NXG.0000000000000015
Expanding genotype/phenotype of neuromuscular diseases by comprehensive target capture/NGS
Abstract
Objective: To establish and evaluate the effectiveness of a comprehensive next-generation sequencing (NGS) approach to simultaneously analyze all genes known to be responsible for the most clinically and genetically heterogeneous neuromuscular diseases (NMDs) involving spinal motoneurons, neuromuscular junctions, nerves, and muscles.
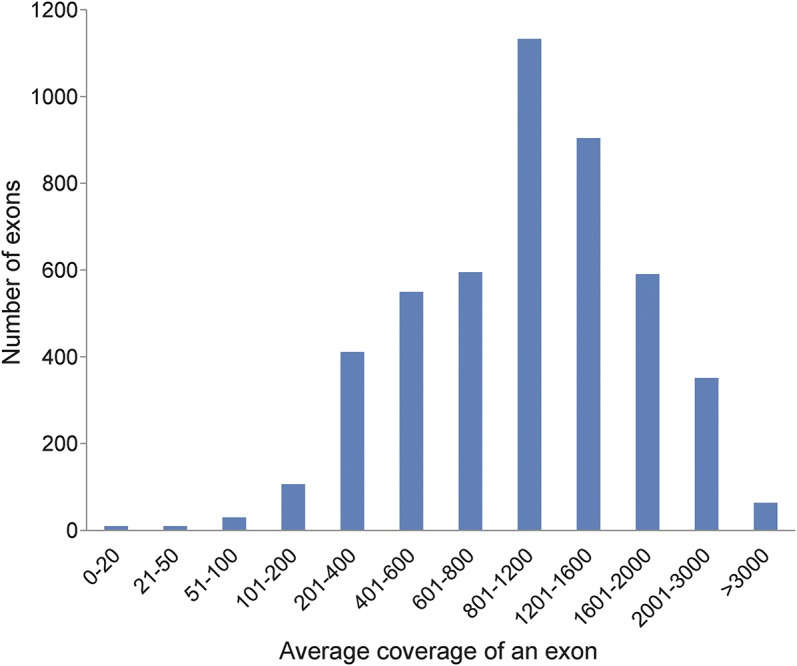
Methods: All coding exons and at least 20 bp of flanking intronic sequences of 236 genes causing NMDs were enriched by using SeqCap EZ solution-based capture and enrichment method followed by massively parallel sequencing on Illumina HiSeq2000.
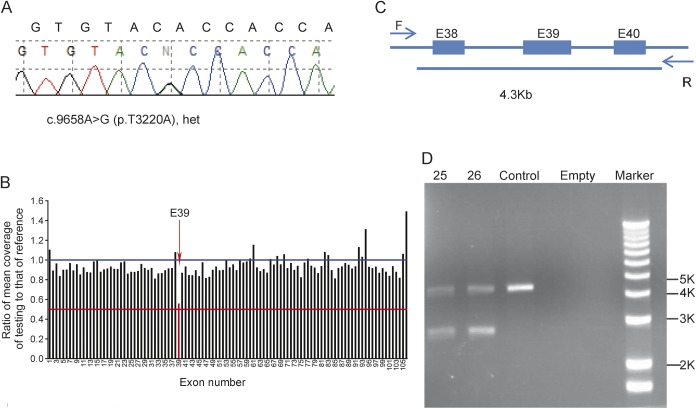
Results: The target gene capture/deep sequencing provides an average coverage of ∼1,000× per nucleotide. Thirty-five unrelated NMD families (38 patients) with clinical and/or muscle pathologic diagnoses but without identified causative genetic defects were analyzed. Deleterious mutations were found in 29 families (83%). Definitive causative mutations were identified in 21 families (60%) and likely diagnoses were established in 8 families (23%). Six families were left without diagnosis due to uncertainty in phenotype/genotype correlation and/or unidentified causative genes. Using this comprehensive panel, we not only identified mutations in expected genes but also expanded phenotype/genotype among different subcategories of NMDs.
Conclusions: Target gene capture/deep sequencing approach can greatly improve the genetic diagnosis of NMDs. This study demonstrated the power of NGS in confirming and expanding clinical phenotypes/genotypes of the extremely heterogeneous NMDs. Confirmed molecular diagnoses of NMDs can assist in genetic counseling and carrier detection as well as guide therapeutic options for treatable disorders.
Figures
Similar articles
-
Dependable and efficient clinical utility of target capture-based deep sequencing in molecular diagnosis of retinitis pigmentosa.Invest Ophthalmol Vis Sci. 2014 Aug 5;55(10):6213-23. doi: 10.1167/iovs.14-14936. Invest Ophthalmol Vis Sci. 2014. PMID: 25097241
-
A comprehensive genomic approach for neuromuscular diseases gives a high diagnostic yield.Ann Neurol. 2015 Feb;77(2):206-14. doi: 10.1002/ana.24303. Epub 2014 Dec 17. Ann Neurol. 2015. PMID: 25380242
-
Target resequencing of neuromuscular disease-related genes using next-generation sequencing for patients with undiagnosed early-onset neuromuscular disorders.J Hum Genet. 2016 Nov;61(11):931-942. doi: 10.1038/jhg.2016.79. Epub 2016 Jun 30. J Hum Genet. 2016. PMID: 27357428
-
Facilitations and Hurdles of Genetic Testing in Neuromuscular Disorders.Diagnostics (Basel). 2021 Apr 14;11(4):701. doi: 10.3390/diagnostics11040701. Diagnostics (Basel). 2021. PMID: 33919863 Free PMC article. Review.
-
Impacts of massively parallel sequencing for genetic diagnosis of neuromuscular disorders.Acta Neuropathol. 2013 Feb;125(2):173-85. doi: 10.1007/s00401-012-1072-7. Epub 2012 Dec 7. Acta Neuropathol. 2013. PMID: 23224362 Review.
Cited by
-
Profiling of pathogenic variants in Japanese patients with sarcoglycanopathy.Orphanet J Rare Dis. 2025 Jan 4;20(1):1. doi: 10.1186/s13023-024-03521-2. Orphanet J Rare Dis. 2025. PMID: 39755676 Free PMC article.
-
Clinical utility of multigene analysis in over 25,000 patients with neuromuscular disorders.Neurol Genet. 2020 Mar 9;6(2):e412. doi: 10.1212/NXG.0000000000000412. eCollection 2020 Apr. Neurol Genet. 2020. PMID: 32337338 Free PMC article.
-
Next generation sequencing panel as an effective approach to genetic testing in patients with a highly variable phenotype of neuromuscular disorders.Neurogenetics. 2024 Jul;25(3):233-247. doi: 10.1007/s10048-024-00762-y. Epub 2024 May 17. Neurogenetics. 2024. PMID: 38758368 Free PMC article.
-
Using gene panels in the diagnosis of neuromuscular disorders: A mini-review.Front Neurol. 2022 Oct 12;13:997551. doi: 10.3389/fneur.2022.997551. eCollection 2022. Front Neurol. 2022. PMID: 36313509 Free PMC article. Review.
-
Clinical, pathological, imaging, and genetic characterization in a Taiwanese cohort with limb-girdle muscular dystrophy.Orphanet J Rare Dis. 2020 Jun 23;15(1):160. doi: 10.1186/s13023-020-01445-1. Orphanet J Rare Dis. 2020. PMID: 32576226 Free PMC article.
References
-
- Kaplan JC, Hamroun D. The 2014 version of the gene table of monogenic neuromuscular disorders (nuclear genome). Neuromuscul Disord 2013;23:1081–1111. - PubMed
-
- Prasad AN, Prasad C. Genetic evaluation of the floppy infant. Semin Fetal Neonatal Med 2011;16:99–108. - PubMed
-
- Laing NG. Genetics of neuromuscular disorders. Crit Rev Clin Lab Sci 2012;49:33–48. - PubMed
-
- Singh RR, Tan SV, Hanna MG, Robb SA, Clarke A, Jungbluth H. Mutations in SCN4A: a rare but treatable cause of recurrent life-threatening laryngospasm. Pediatrics 2014;134:e1447–e1450. - PubMed
LinkOut - more resources
Full Text Sources