

Mutation in PNKP presenting initially as axonal Charcot-Marie-Tooth disease
- PMID: 27066567
- PMCID: PMC4811384
- DOI: 10.1212/NXG.0000000000000030
Mutation in PNKP presenting initially as axonal Charcot-Marie-Tooth disease
Abstract
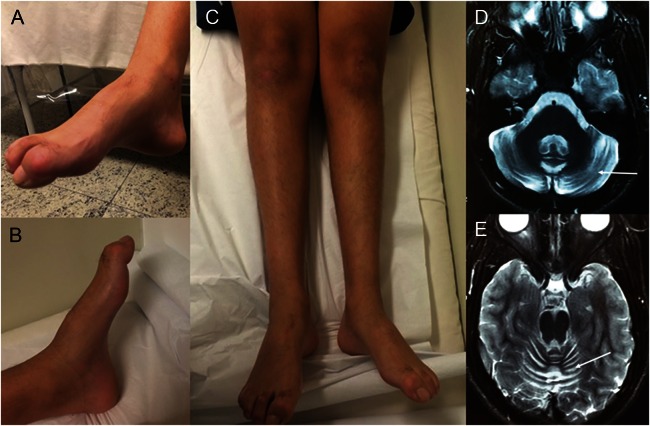
PNKP (polynucleotide kinase 3'-phosphatase, OMIM #605610) product is involved in the repair of strand breaks and base damage in the DNA molecule mainly caused by radical oxygen species. Deleterious variants affecting this gene have been previously associated with microcephaly, epilepsy, and developmental delay.(1) According to a previous report, homozygous loss-of-function substitution in PNKP was associated with cerebellar atrophy, neuropathy, microcephaly, epilepsy, and intellectual disability.(2) Recently, whole-exome sequencing (WES) performed in a cohort of Portuguese families with ataxia with oculomotor apraxia (AOA) disclosed pathogenic variants in PNKP in 11 individuals. Other clinical features in that study included neuropathy, dystonia, cognitive impairment, decreased vibration sense, pyramidal signs, mild elevation in α-fetoprotein, and low levels of albumin. This condition was named AOA type 4 (OMIM #616267), as the phenotype of AOA has been previously associated with 3 other genes: APTX, SETX, and PIK3R5.(3) Altogether, these reports demonstrate the great phenotypic diversity associated with PNKP mutations. In this article, we further enlarge this variability by demonstrating that early-onset axonal sensory-motor neuropathy (or axonal Charcot-Marie-Tooth (CMT) disease) followed years later by ataxia without oculomotor apraxia can be caused by deleterious variants in PNKP. Full consent was obtained from the patient and his parents for this publication. This study was approved by institutional ethics committees.
Figures
Similar articles
-
The polynucleotide kinase 3'-phosphatase gene (PNKP) is involved in Charcot-Marie-Tooth disease (CMT2B2) previously related to MED25.Neurogenetics. 2018 Dec;19(4):215-225. doi: 10.1007/s10048-018-0555-7. Epub 2018 Jul 24. Neurogenetics. 2018. PMID: 30039206 Free PMC article.
-
From congenital microcephaly to adult onset cerebellar ataxia: Distinct and overlapping phenotypes in patients with PNKP gene mutations.Am J Med Genet A. 2019 Nov;179(11):2277-2283. doi: 10.1002/ajmg.a.61339. Epub 2019 Aug 22. Am J Med Genet A. 2019. PMID: 31436889
-
The Phenotypic Spectrum of PNKP-Associated Disease and the Absence of Immunodeficiency and Cancer Predisposition in a Dutch Cohort.Pediatr Neurol. 2020 Dec;113:26-32. doi: 10.1016/j.pediatrneurol.2020.07.014. Epub 2020 Jul 28. Pediatr Neurol. 2020. PMID: 32980744
-
Polynucleotide kinase-phosphatase (PNKP) mutations and neurologic disease.Mech Ageing Dev. 2017 Jan;161(Pt A):121-129. doi: 10.1016/j.mad.2016.04.009. Epub 2016 Apr 26. Mech Ageing Dev. 2017. PMID: 27125728 Free PMC article. Review.
-
[Molecular genetics of inherited neuropathies].Rinsho Shinkeigaku. 2006 Jan;46(1):1-18. Rinsho Shinkeigaku. 2006. PMID: 16541790 Review. Japanese.
Cited by
-
Late-Onset Friedreich's Ataxia (LOFA) Mimicking Charcot-Marie-Tooth Disease Type 2: What Is Similar and What Is Different?Cerebellum. 2017 Apr;16(2):599-601. doi: 10.1007/s12311-016-0822-9. Cerebellum. 2017. PMID: 27687732
-
Pathological mutations in PNKP trigger defects in DNA single-strand break repair but not DNA double-strand break repair.Nucleic Acids Res. 2020 Jul 9;48(12):6672-6684. doi: 10.1093/nar/gkaa489. Nucleic Acids Res. 2020. PMID: 32504494 Free PMC article.
-
Ataxia with Oculomotor Apraxia Type 4 with PNKP Common "Portuguese" and Novel Mutations in Two Belarusian Families.J Pediatr Genet. 2019 Jun;8(2):58-62. doi: 10.1055/s-0039-1684008. Epub 2019 Mar 27. J Pediatr Genet. 2019. PMID: 31061747 Free PMC article.
-
Novel PNKP mutations causing defective DNA strand break repair and PARP1 hyperactivity in MCSZ.Neurol Genet. 2019 Mar 25;5(2):e320. doi: 10.1212/NXG.0000000000000320. eCollection 2019 Apr. Neurol Genet. 2019. PMID: 31041400 Free PMC article.
-
A decade of whole-exome sequencing in Brazilian Neurology: from past insights to future perspectives.Arq Neuropsiquiatr. 2025 Apr;83(4):1-14. doi: 10.1055/s-0045-1807715. Epub 2025 May 13. Arq Neuropsiquiatr. 2025. PMID: 40360003 Free PMC article. Review.
References
-
- Poulton C, Oegema R, Heijsman D, et al. Progressive cerebellar atrophy and polyneuropathy: expanding the spectrum of PNKP mutations. Neurogenetics 2013;14:43–51. - PubMed
-
- Cleaver JE. γH2Ax: biomarker of damage or functional Participant in DNA repair “all that glitters is not g old!” Photochem Photobiol 2011;87:1230–1239. - PubMed
LinkOut - more resources
Full Text Sources
Molecular Biology Databases