

Identifying contamination with advanced visualization and analysis practices: metagenomic approaches for eukaryotic genome assemblies
- PMID: 27069789
- PMCID: PMC4824900
- DOI: 10.7717/peerj.1839
Identifying contamination with advanced visualization and analysis practices: metagenomic approaches for eukaryotic genome assemblies
Abstract
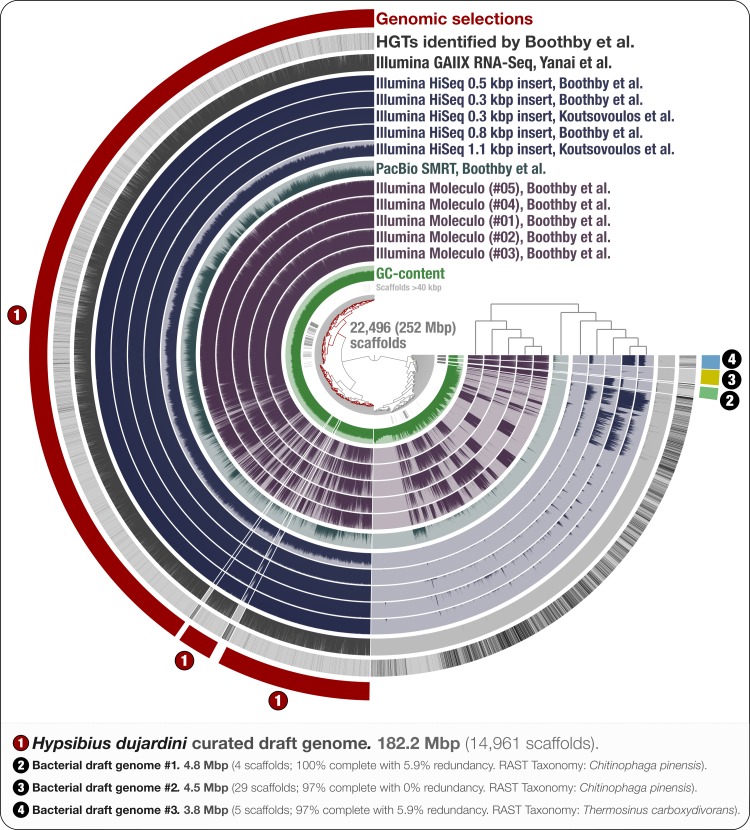
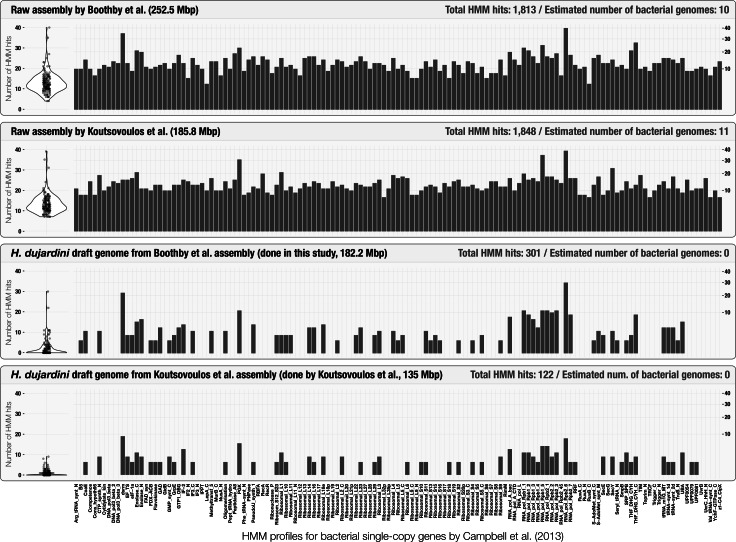
High-throughput sequencing provides a fast and cost-effective mean to recover genomes of organisms from all domains of life. However, adequate curation of the assembly results against potential contamination of non-target organisms requires advanced bioinformatics approaches and practices. Here, we re-analyzed the sequencing data generated for the tardigrade Hypsibius dujardini, and created a holistic display of the eukaryotic genome assembly using DNA data originating from two groups and eleven sequencing libraries. By using bacterial single-copy genes, k-mer frequencies, and coverage values of scaffolds we could identify and characterize multiple near-complete bacterial genomes from the raw assembly, and curate a 182 Mbp draft genome for H. dujardini supported by RNA-Seq data. Our results indicate that most contaminant scaffolds were assembled from Moleculo long-read libraries, and most of these contaminants have differed between library preparations. Our re-analysis shows that visualization and curation of eukaryotic genome assemblies can benefit from tools designed to address the needs of today's microbiologists, who are constantly challenged by the difficulties associated with the identification of distinct microbial genomes in complex environmental metagenomes.
Keywords: Assembly; Contamination; Curation; Genomics; HGT; Visualization.
Conflict of interest statement
A. Murat Eren is an Academic Editor for PeerJ.
Figures
Comment in
-
Reply to Bemm et al. and Arakawa: Identifying foreign genes in independent Hypsibius dujardini genome assemblies.Proc Natl Acad Sci U S A. 2016 May 31;113(22):E3058-61. doi: 10.1073/pnas.1601149113. Epub 2016 May 12. Proc Natl Acad Sci U S A. 2016. PMID: 27173900 Free PMC article. No abstract available.
-
No evidence for extensive horizontal gene transfer from the draft genome of a tardigrade.Proc Natl Acad Sci U S A. 2016 May 31;113(22):E3057. doi: 10.1073/pnas.1602711113. Epub 2016 May 12. Proc Natl Acad Sci U S A. 2016. PMID: 27173901 Free PMC article. No abstract available.
References
-
- Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA, Formsma K, Gerdes S, Glass EM, Kubal M, Meyer F, Olsen GJ, Olson R, Osterman AL, Overbeek RA, McNeil LK, Paarmann D, Paczian T, Parrello B, Pusch GD, Reich C, Stevens R, Vassieva O, Vonstein V, Wilke A, Zagnitko O. The RAST Server: rapid annotations using subsystems technology. BMC Genomics. 2008;9:75. doi: 10.1186/1471-2164-9-75. - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
