

A Comprehensive Analysis of Choroideremia: From Genetic Characterization to Clinical Practice
- PMID: 27070432
- PMCID: PMC4829155
- DOI: 10.1371/journal.pone.0151943
A Comprehensive Analysis of Choroideremia: From Genetic Characterization to Clinical Practice
Abstract
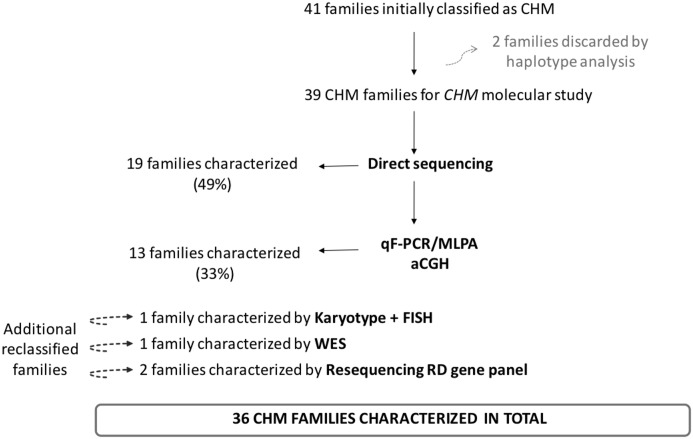
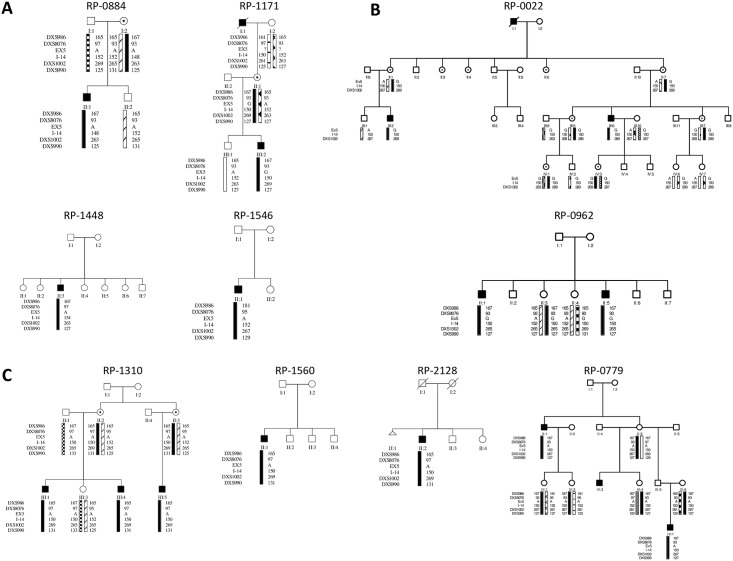
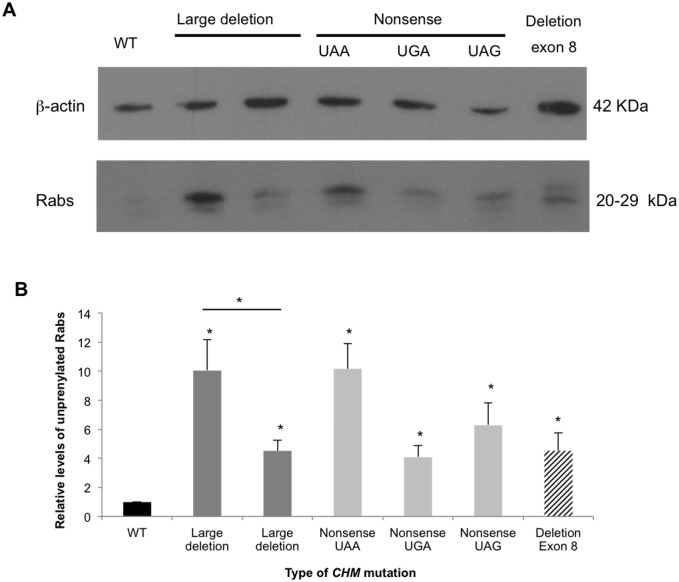
Choroideremia (CHM) is a rare X-linked disease leading to progressive retinal degeneration resulting in blindness. The disorder is caused by mutations in the CHM gene encoding REP-1 protein, an essential component of the Rab geranylgeranyltransferase (GGTase) complex. In the present study, we evaluated a multi-technique analysis algorithm to describe the mutational spectrum identified in a large cohort of cases and further correlate CHM variants with phenotypic characteristics and biochemical defects of choroideremia patients. Molecular genetic testing led to the characterization of 36 out of 45 unrelated CHM families (80%), allowing the clinical reclassification of four CHM families. Haplotype reconstruction showed independent origins for the recurrent p.Arg293* and p.Lys178Argfs*5 mutations, suggesting the presence of hotspots in CHM, as well as the identification of two different unrelated events involving exon 9 deletion. No certain genotype-phenotype correlation could be established. Furthermore, all the patients´ fibroblasts analyzed presented significantly increased levels of unprenylated Rabs proteins compared to control cells; however, this was not related to the genotype. This research demonstrates the major potential of the algorithm proposed for diagnosis. Our data enhance the importance of establish a differential diagnosis with other retinal dystrophies, supporting the idea of an underestimated prevalence of choroideremia. Moreover, they suggested that the severity of the disorder cannot be exclusively explained by the genotype.
Conflict of interest statement
Figures
References
-
- MacDonald IM, Hume S, Chan S, Seabra MC. Choroideremia In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, et al. , editors. GeneReviews® [Internet] Seattle (WA): University of Washington, Seattle; 1993–2015. 2003. Epub 2010/03/20. doi: NBK1337 [bookaccession]. .
-
- Seabra MC, Brown MS, Slaughter CA, Sudhof TC, Goldstein JL. Purification of component A of Rab geranylgeranyl transferase: possible identity with the choroideremia gene product. Cell. 1992;70(6):1049–57. Epub 1992/09/18. doi: 0092-8674(92)90253-9 [pii]. . - PubMed
-
- Seabra MC, Brown MS, Goldstein JL. Retinal degeneration in choroideremia: deficiency of rab geranylgeranyl transferase. Science. 1993;259(5093):377–81. Epub 1993/01/15. . - PubMed
-
- Andres DA, Seabra MC, Brown MS, Armstrong SA, Smeland TE, Cremers FP, et al. cDNA cloning of component A of Rab geranylgeranyl transferase and demonstration of its role as a Rab escort protein. Cell. 1993;73(6):1091–9. Epub 1993/06/18. doi: 0092-8674(93)90639-8 [pii]. . - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
