

Mapping physiological G protein-coupled receptor signaling pathways reveals a role for receptor phosphorylation in airway contraction
- PMID: 27071102
- PMCID: PMC4843461
- DOI: 10.1073/pnas.1521706113
Mapping physiological G protein-coupled receptor signaling pathways reveals a role for receptor phosphorylation in airway contraction
Abstract
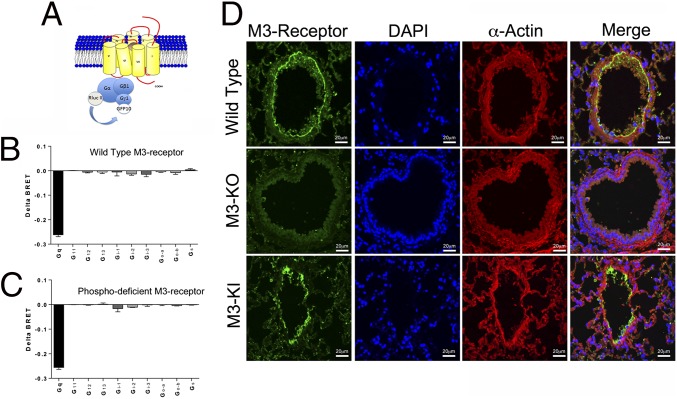
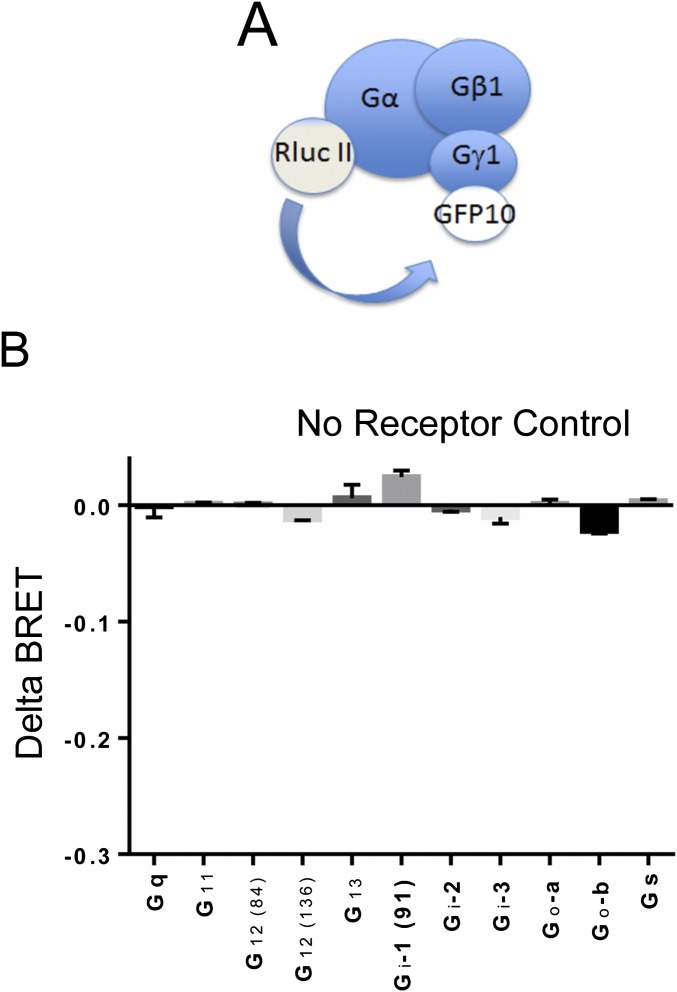
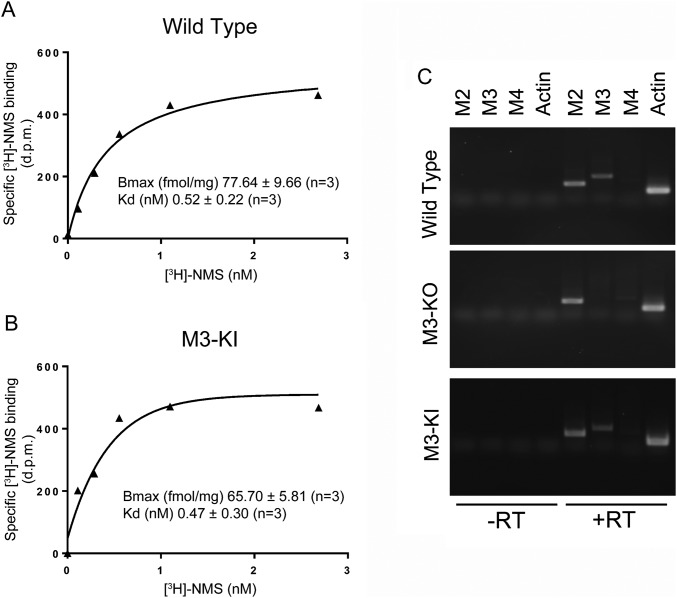
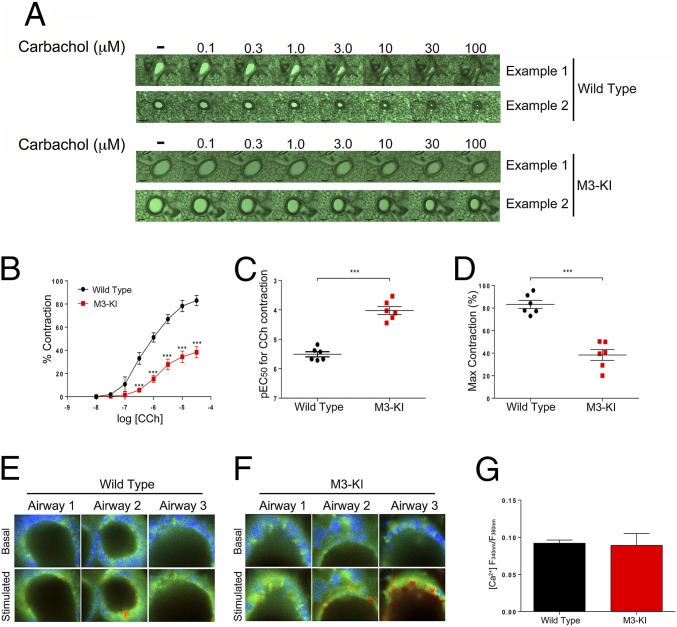
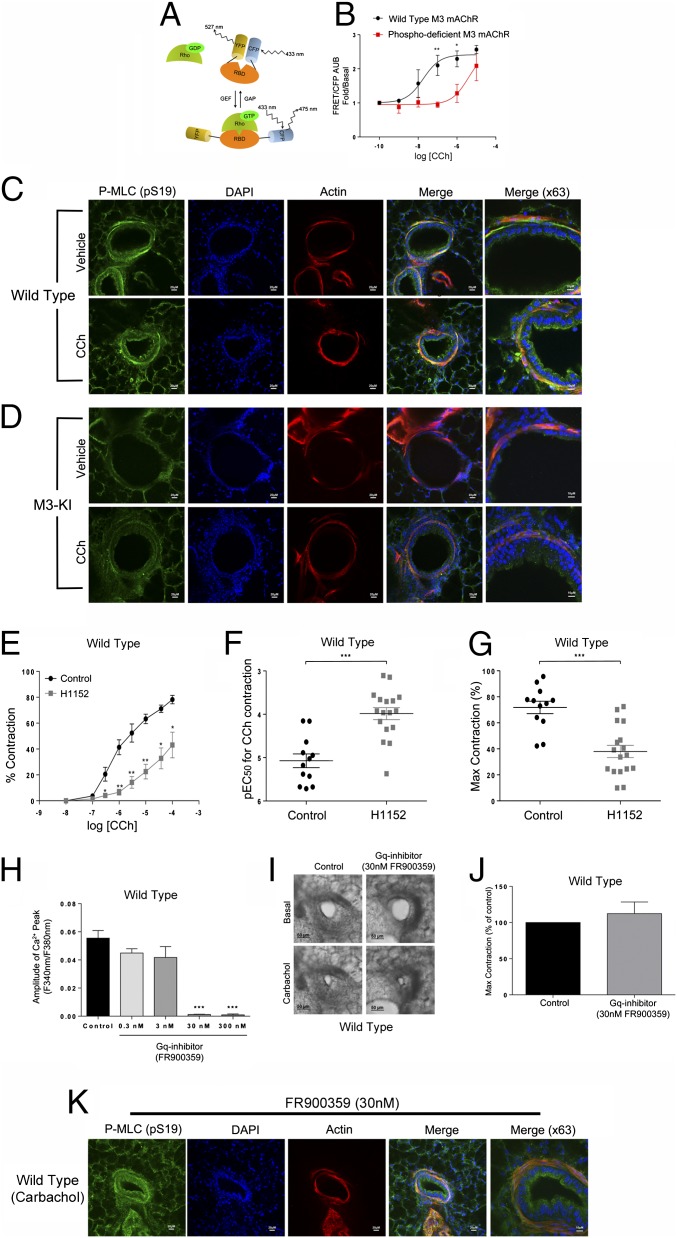
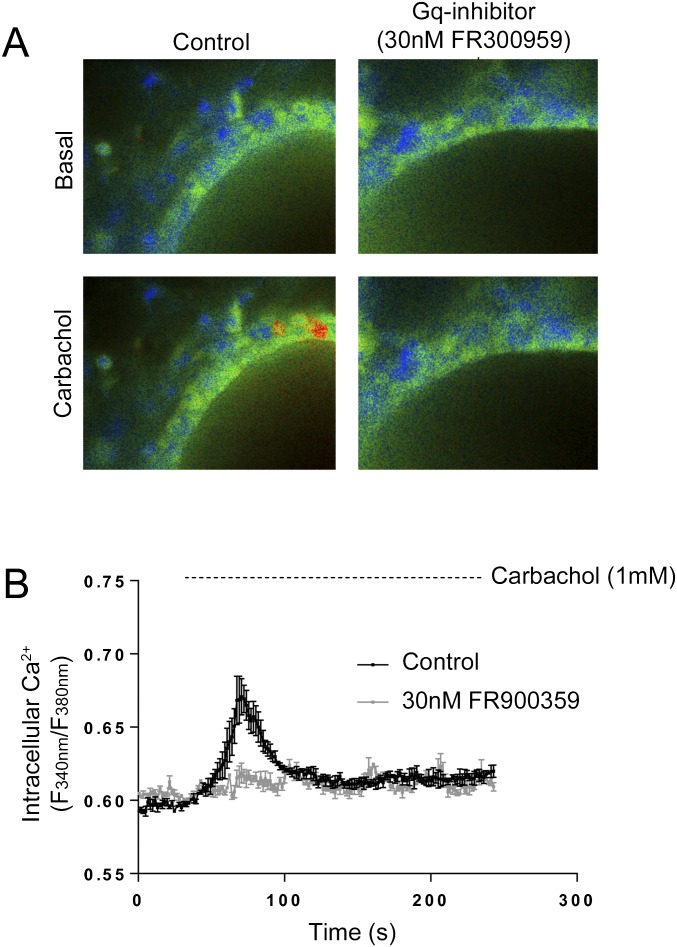
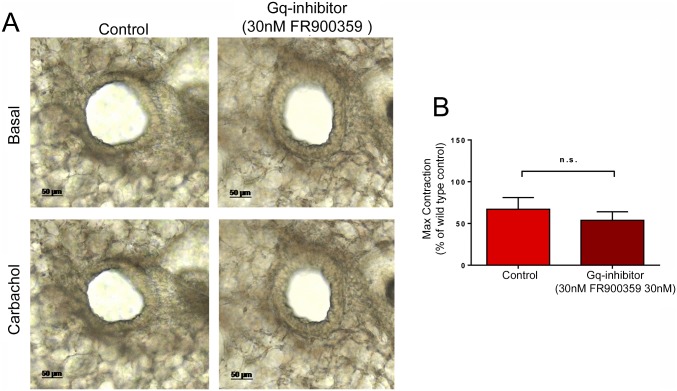
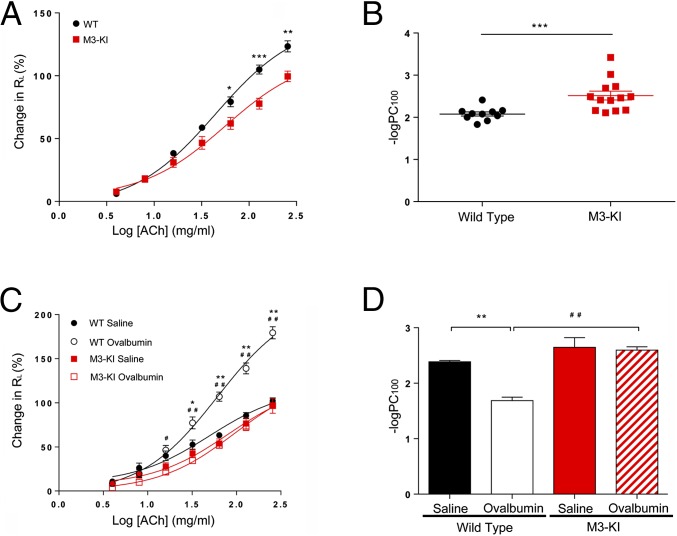
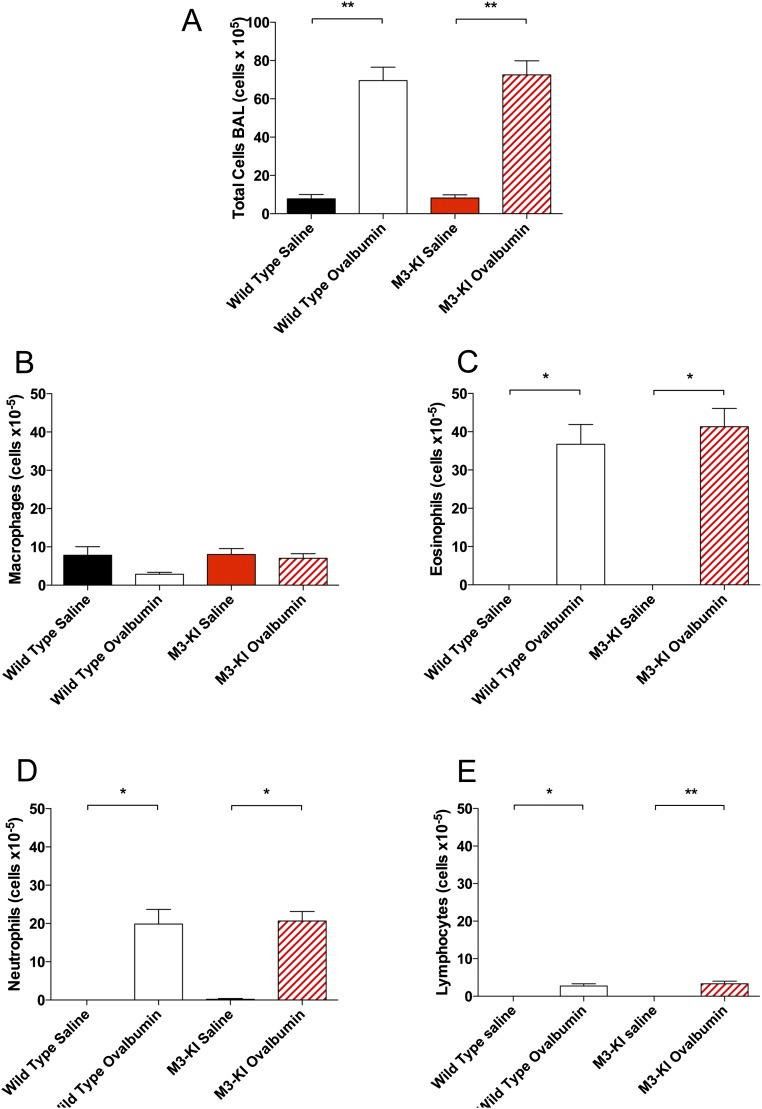
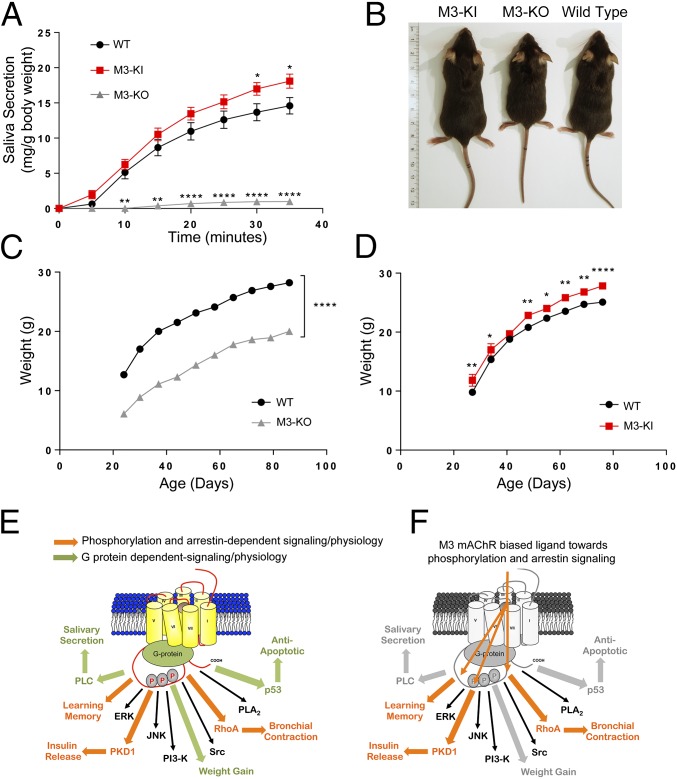
G protein-coupled receptors (GPCRs) are known to initiate a plethora of signaling pathways in vitro. However, it is unclear which of these pathways are engaged to mediate physiological responses. Here, we examine the distinct roles of Gq/11-dependent signaling and receptor phosphorylation-dependent signaling in bronchial airway contraction and lung function regulated through the M3-muscarinic acetylcholine receptor (M3-mAChR). By using a genetically engineered mouse expressing a G protein-biased M3-mAChR mutant, we reveal the first evidence, to our knowledge, of a role for M3-mAChR phosphorylation in bronchial smooth muscle contraction in health and in a disease state with relevance to human asthma. Furthermore, this mouse model can be used to distinguish the physiological responses that are regulated by M3-mAChR phosphorylation (which include control of lung function) from those responses that are downstream of G protein signaling. In this way, we present an approach by which to predict the physiological/therapeutic outcome of M3-mAChR-biased ligands with important implications for drug discovery.
Keywords: G protein-coupled receptor; asthma; ligand bias; muscarinic; signaling.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Somlyo AP, Somlyo AV. Ca2+ sensitivity of smooth muscle and nonmuscle myosin II: Modulated by G proteins, kinases, and myosin phosphatase. Physiol Rev. 2003;83(4):1325–1358. - PubMed
-
- Bai Y, Sanderson MJ. Modulation of the Ca2+ sensitivity of airway smooth muscle cells in murine lung slices. Am J Physiol Lung Cell Mol Physiol. 2006;291(2):L208–L221. - PubMed
-
- Liu C, Zuo J, Janssen LJ. Regulation of airway smooth muscle RhoA/ROCK activities by cholinergic and bronchodilator stimuli. Eur Respir J. 2006;28(4):703–711. - PubMed
