

Induction of autoimmune disease by deletion of CTLA-4 in mice in adulthood
- PMID: 27071130
- PMCID: PMC4855592
- DOI: 10.1073/pnas.1603892113
Induction of autoimmune disease by deletion of CTLA-4 in mice in adulthood
Abstract
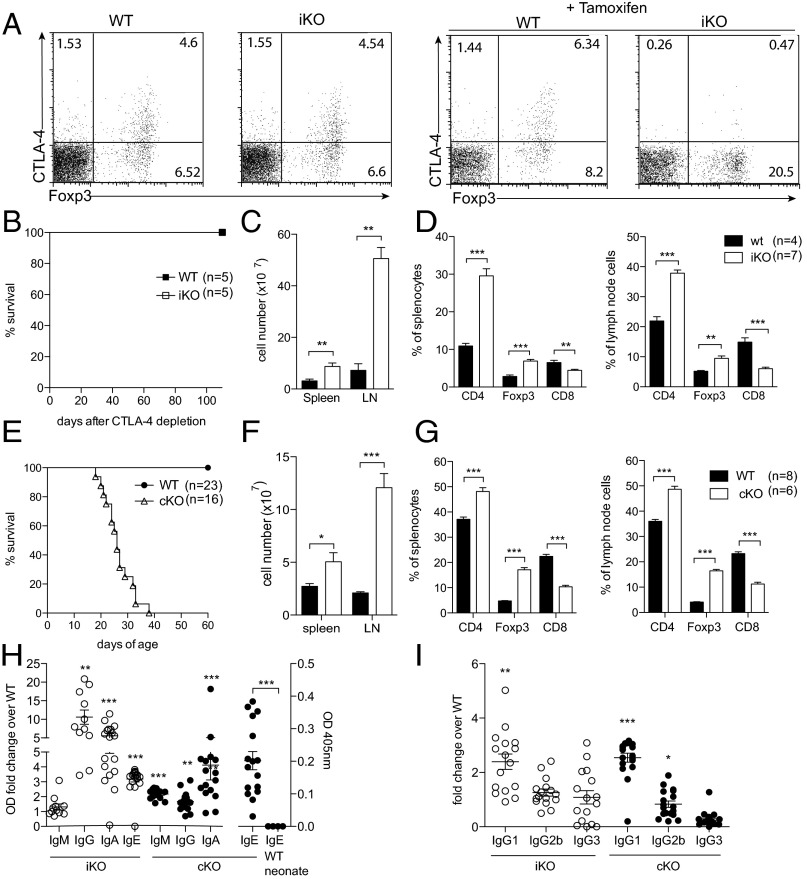
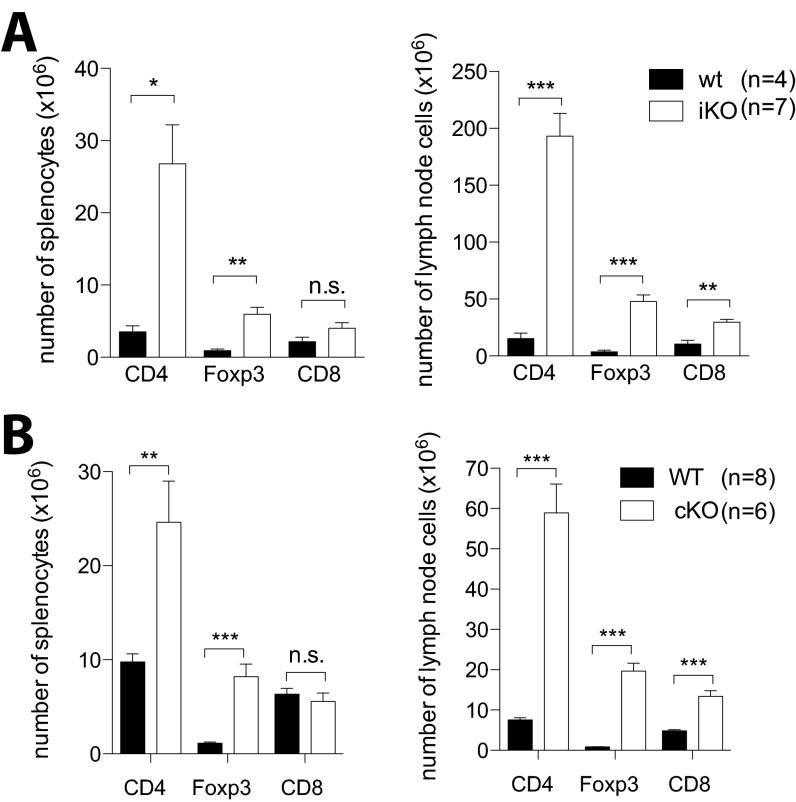
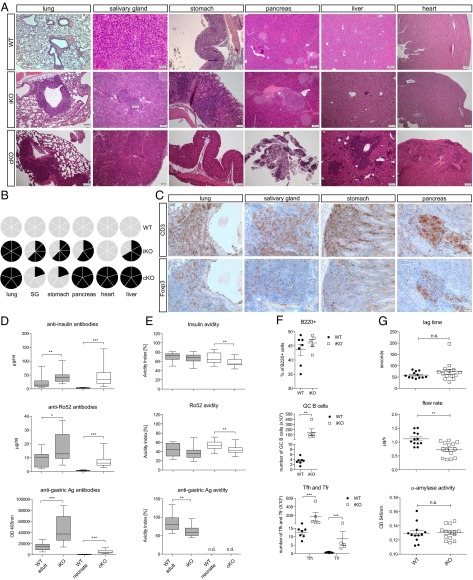
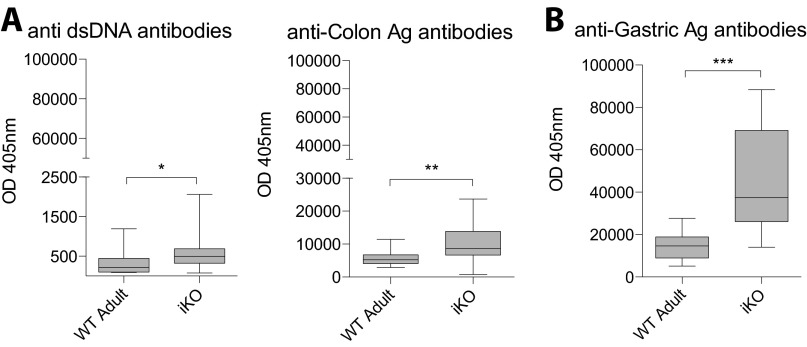
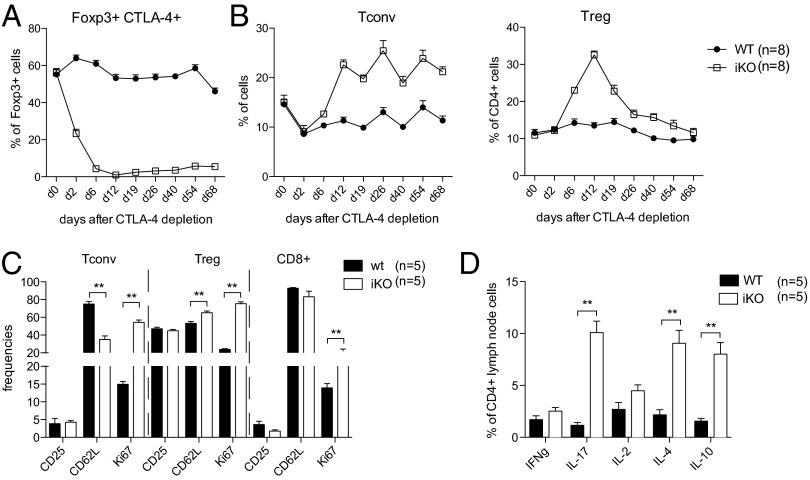
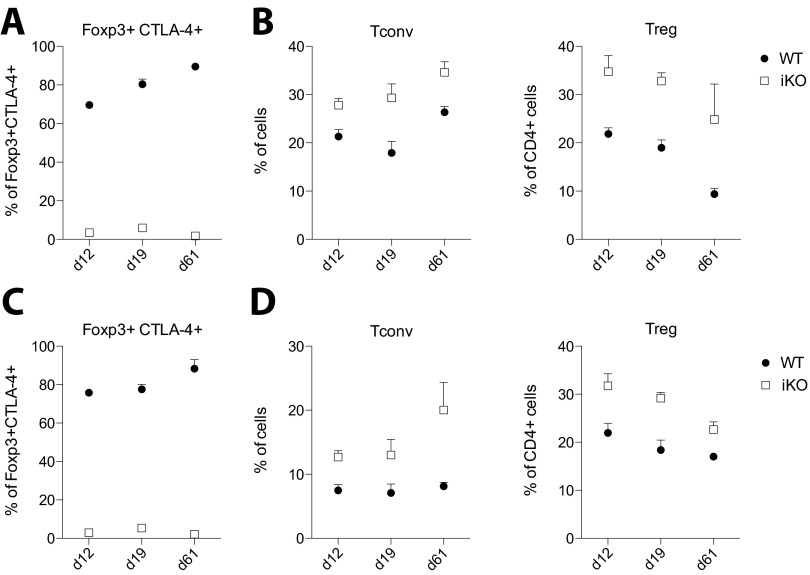
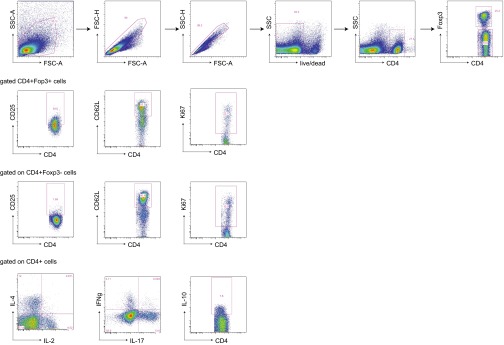
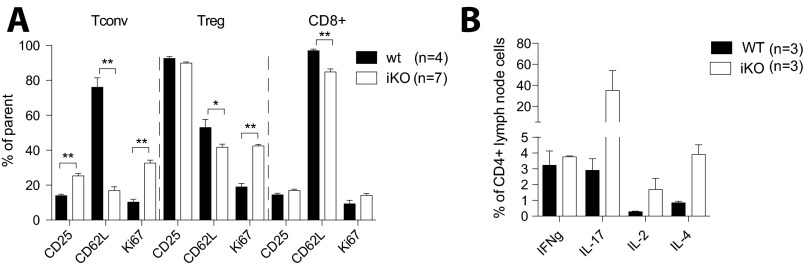
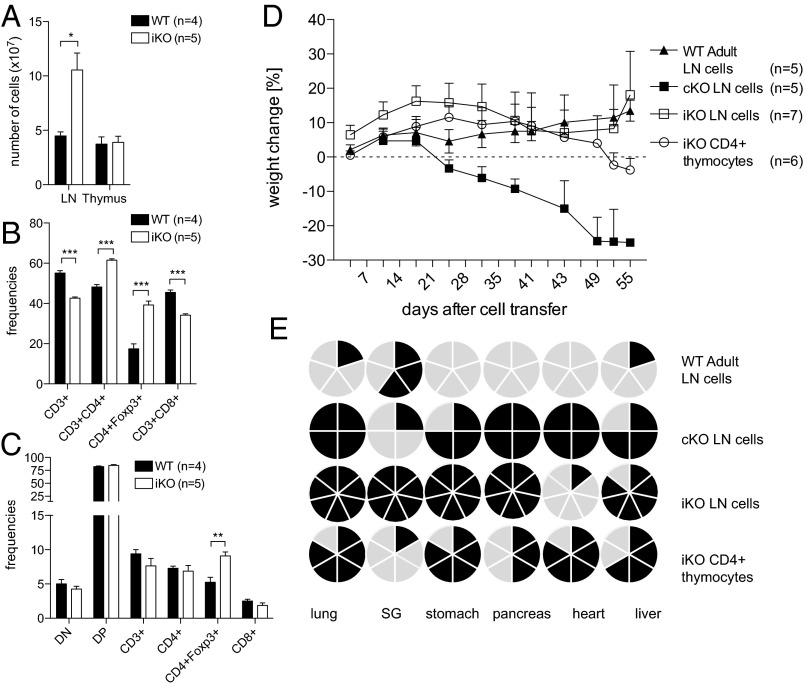
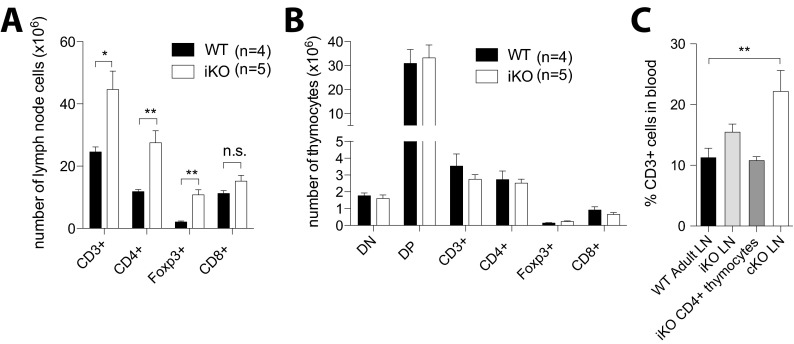
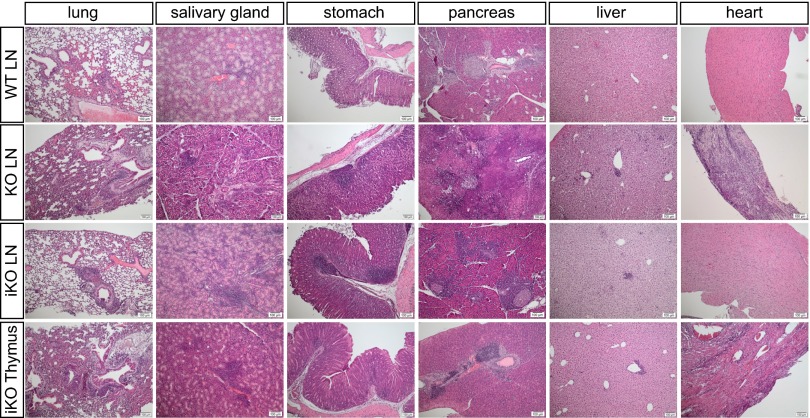
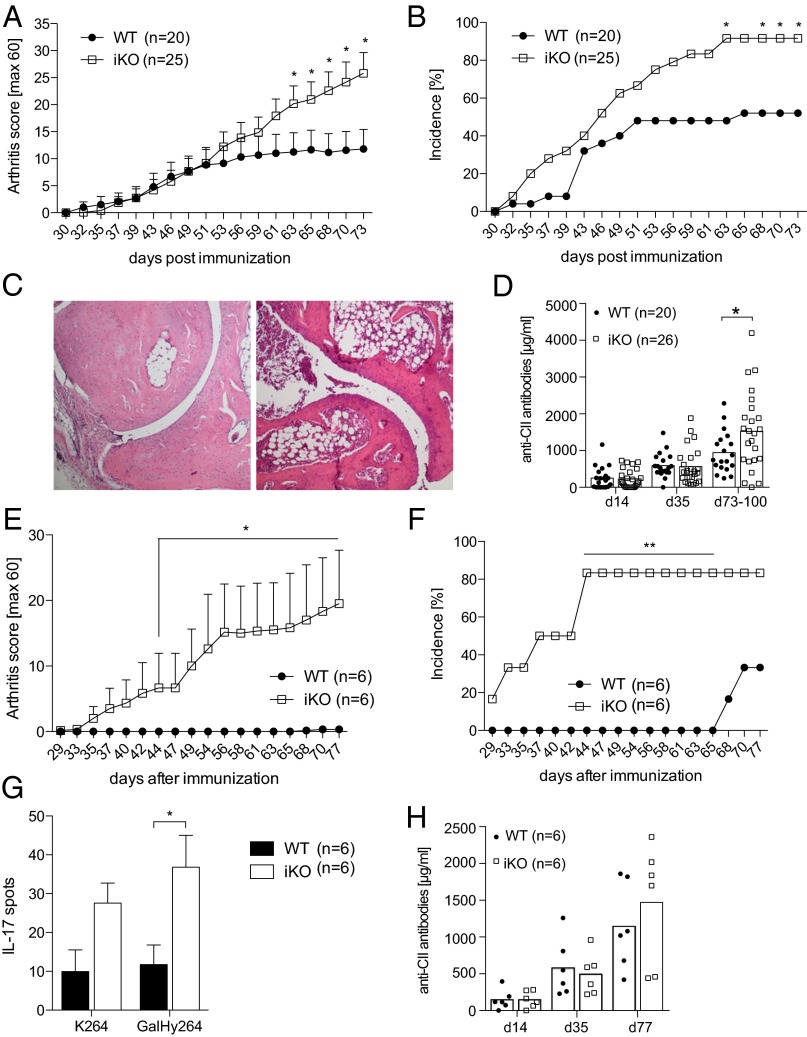
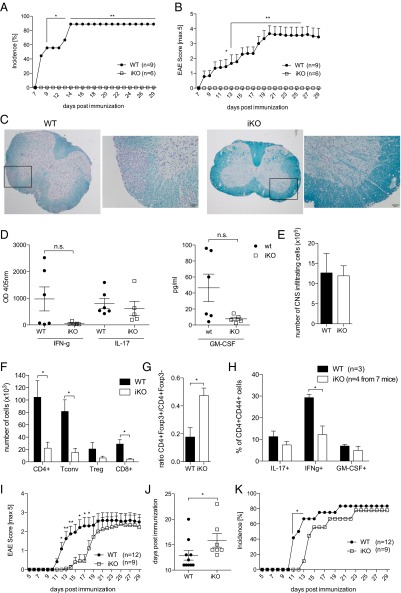
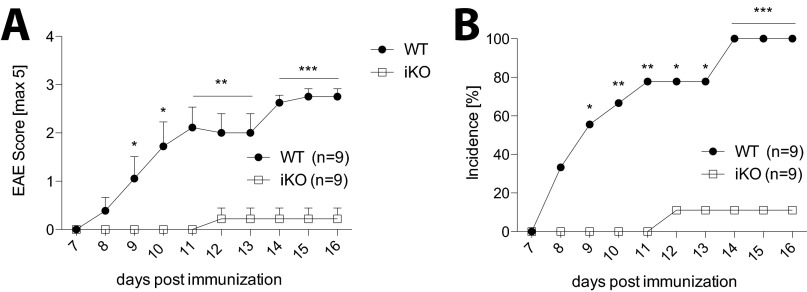
Cytotoxic T lymphocyte antigen-4 (CTLA-4) is essential for immunological (self-) tolerance, but due to the early fatality of CTLA-4 KO mice, its specific function in central and peripheral tolerance and in different systemic diseases remains to be determined. Here, we further examined the role of CTLA-4 by abrogating CTLA-4 expression in adult mice and compared the resulting autoimmunity that follows with that produced by congenital CTLA-4 deficiency. We found that conditional deletion of CTLA-4 in adult mice resulted in spontaneous lymphoproliferation, hypergammaglobulinemia, and histologically evident pneumonitis, gastritis, insulitis, and sialadenitis, accompanied by organ-specific autoantibodies. However, in contrast to congenital deficiency, this was not fatal. CTLA-4 deletion induced preferential expansion of CD4(+)Foxp3(+) Treg cells. However, T cells from CTLA-4-deficient inducible KO mice were able to adoptively transfer the diseases into T cell-deficient mice. Notably, cell transfer of thymocytes de novo produced myocarditis, otherwise not observed in donor mice depleted in adulthood. Moreover, CTLA-4 deletion in adult mice had opposing impacts on induced autoimmune models. Thus, although CTLA-4-deficient mice had more severe collagen-induced arthritis (CIA), they were protected against peptide-induced experimental autoimmune encephalomyelitis (EAE); however, onset of protein-induced EAE was only delayed. Collectively, this indicates that CTLA-4 deficiency affects both central and peripheral tolerance and Treg cell-mediated suppression.
Keywords: CTLA-4; Foxp3; autoimmunity; regulatory T cells; tolerance.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Thompson CB, Allison JP. The emerging role of CTLA-4 as an immune attenuator. Immunity. 1997;7(4):445–450. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
