

Augmenting drug-carrier compatibility improves tumour nanotherapy efficacy
- PMID: 27071376
- PMCID: PMC4833858
- DOI: 10.1038/ncomms11221
Augmenting drug-carrier compatibility improves tumour nanotherapy efficacy
Abstract
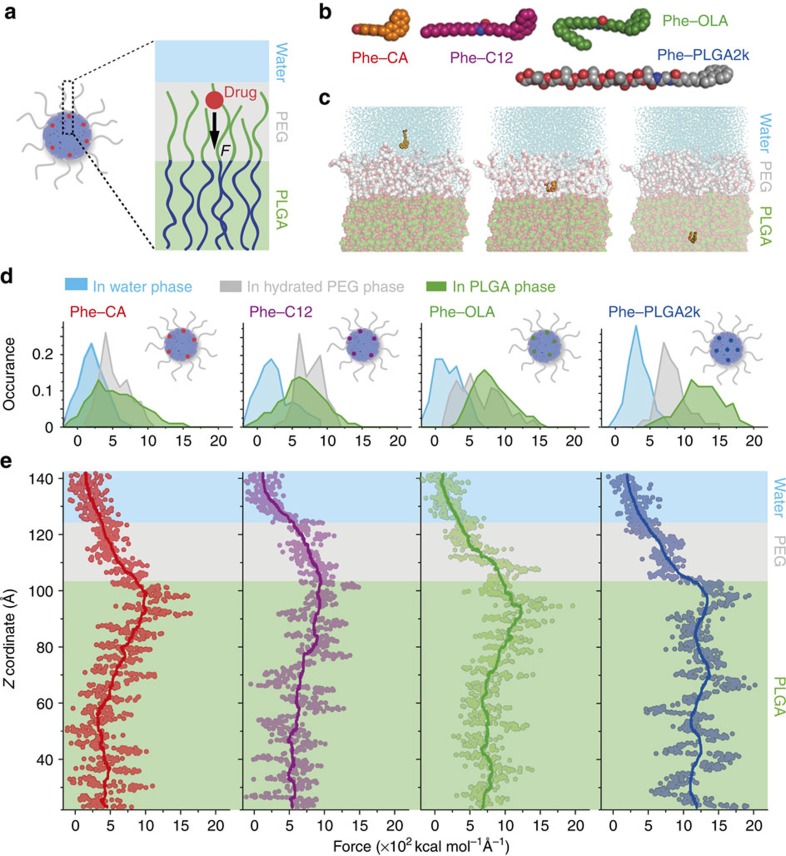
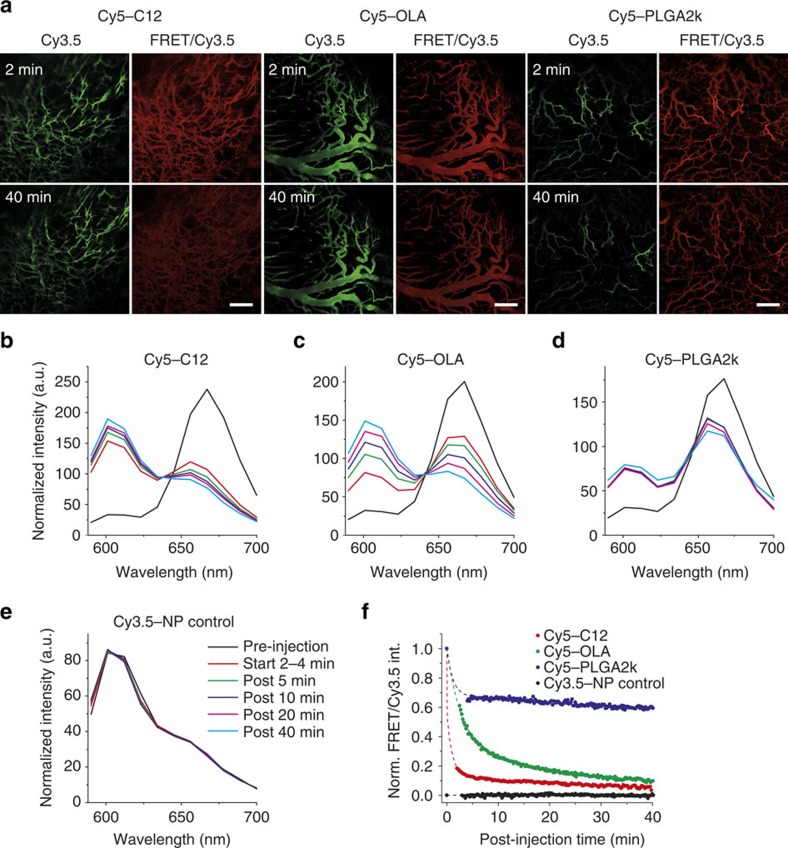
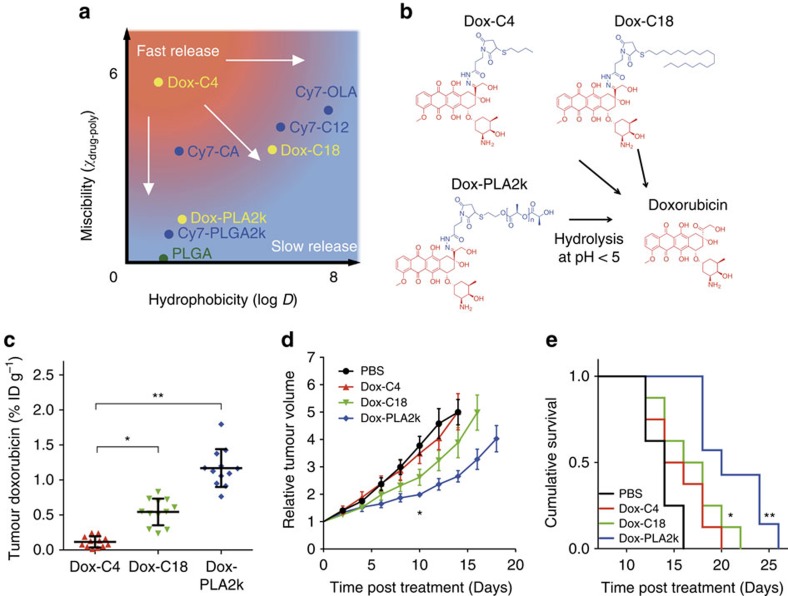
A major goal of cancer nanotherapy is to use nanoparticles as carriers for targeted delivery of anti-tumour agents. The drug-carrier association after intravenous administration is essential for efficient drug delivery to the tumour. However, a large number of currently available nanocarriers are self-assembled nanoparticles whose drug-loading stability is critically affected by the in vivo environment. Here we used in vivo FRET imaging to systematically investigate how drug-carrier compatibility affects drug release in a tumour mouse model. We found the drug's hydrophobicity and miscibility with the nanoparticles are two independent key parameters that determine its accumulation in the tumour. Next, we applied these findings to improve chemotherapeutic delivery by augmenting the parent drug's compatibility; as a result, we achieved better antitumour efficacy. Our results help elucidate nanomedicines' in vivo fate and provide guidelines for efficient drug delivery.
Figures



References
-
- Schroeder A. et al.. Treating metastatic cancer with nanotechnology. Nat. Rev. Cancer 12, 39–50 (2012). - PubMed
-
- Davis M. E., Chen Z. & Shin D. M. Nanoparticle therapeutics: an emerging treatment modality for cancer. Nat. Rev. Drug Discov. 7, 771–782 (2008). - PubMed
-
- Nie S., Xing Y., Kim G. J. & Simons J. W. Nanotechnology applications in cancer. Annu. Rev. Biomed. Eng. 9, 257–288 (2007). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
